Атрофия мышц что это такое: Атрофия мышц — признаки, причины, симптомы, лечение и профилактика
Спинальная амиотрофия I, II, III, IV: генетическая диагностика
Спинальная мышечная атрофия (СМА) или проксимальная спинальная амиотрофия – это наследственное заболевание, при котором происходит нарушение функции нервных клеток спинного мозга, приводящее к прогрессивному развитию слабости мышц, их атрофии, и в итоге, обездвиживанию пациента.Каждый 35 человек является бессимптомным носителем мутации, приводящей к СМА, и больной ребенок рождается, когда встречаются 2 таких мутации, со стороны матери и со стороны отца. Это происходит примерно 1 раз на 6 000 рождений — в семьях, где никто, как правило, не слышал про такую болезнь, где не было больных родственников, вредных факторов среды — ничего, что могло бы навести на мысль о высоком риске генетических проблем.
Причиной заболевания являются мутации в гене SMN1, расположенном на 5 хромосоме.
Поскольку это заболевание является наследственным, для постановки верного диагноза важно, чтобы врач-генетик подробно собрал семейный анамнез и назначил необходимое генетическое тестирование.
В ходе анализа проверяется количество копий 7 и 8 экзонов в генах SMN1 и SMN2.
Важно! При подтверждении диагноза в семье необходимо провести генетическое обследование близких родственников пациента.
К сожалению, на сегодняшний день специфического лечения заболевания не существует. Возможна лишь симптоматическая терапия: физиотерапия, массаж и др.
Для семей, которые уже столкнулись с этим заболеванием и хотят в дальнейшем иметь здоровых детей, существует несколько вариантов профилактики: проведение пренатальной диагностики, использование донорской спермы и яйцеклеток, преимплантационная генетическая диагностика (ПГД). ПГД позволяет обследовать эмбрионы, полученные при ЭКО, еще до беременности и выбрать тот, что не унаследовал заболевание. Все больше семей в мире предпочитают именно такой формат профилактики.
Некоторые семьи выбирают отказ от рождения детей и усыновление.
Узнать о риске, не дожидаясь рождения больного ребенка, можно с помощью генетического скрининга, разработанного лабораторией Genetico, на носительство мутаций, приводящих к СМА.
Лечение спинальной мышечной атрофии (CMA)
В больнице «Ихилов» теперь предоставляется новаторская терапия СМА (спинальной мышечной атрофии) — заболевания, которое до сих пор считалось неизлечимым. Речь идет о лечении препаратом Спинраза (Spinraza). Лекарство получило утверждение Управления по контролю качества пищевых продуктов и лекарственных средств в США.
Спинраза доказала свою эффективность в качестве терапии, спасающей жизнь при СМА. Речь идет о первом препарате, показавшем столь исключительные результаты. Лекарство предотвращает прогрессирование заболевания и улучшает функциональный статус больных. Некоторые из пациентов, получавших Спинразу, вновь обрели способность самостоятельно ходить. Специалисты характеризуют воздействие лекарства как «чудодейственное».
Что такое болезнь Спинальная мышечная атрофия (CMA)?
СМА (спинальная мышечная атрофия) — редкое генетическое заболевание, передающееся наследственным путем в тех случаях, когда оба родителя являются носителями.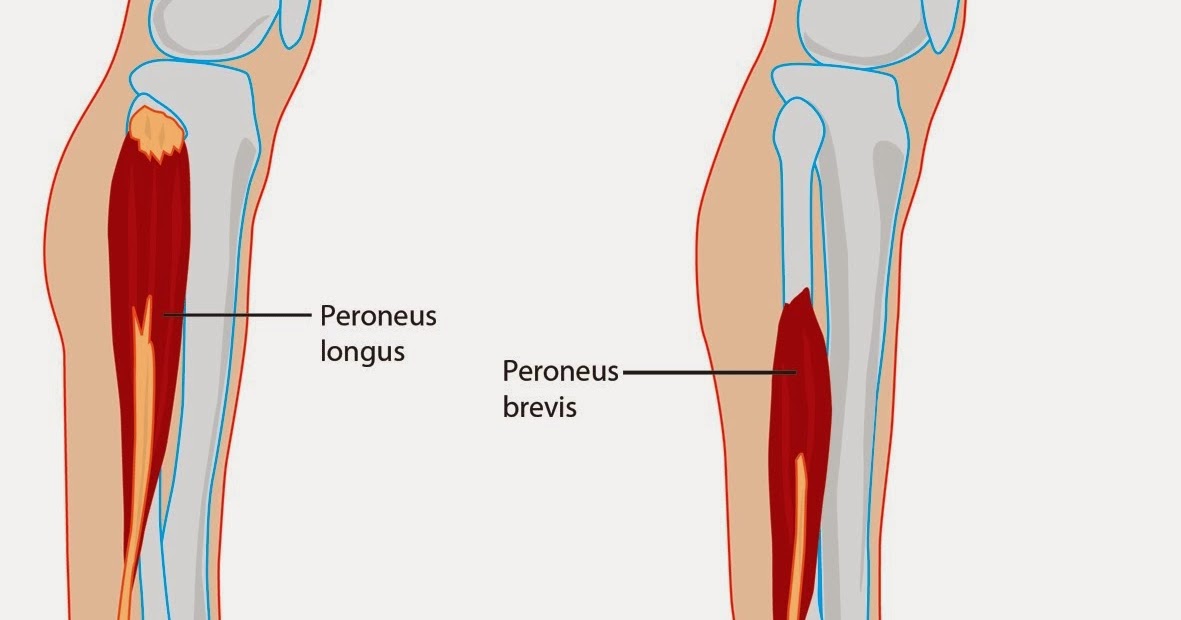 Недуг проявляется в слабости мышц, вплоть до паралича. В большинстве случаев в процесс вовлекаются и дыхательные мышцы. Заболевание развивается вследствие генетического дефекта, приводящего к атрофии определенного рода нервных клеток передних рогов спинного мозга. Встречаемость заболевания составляет от 1:6000 до 1:11000. Диагноз ставится обычно в младенческом или в раннем дошкольном возрасте. Чем раньше появляются первые симптомы, тем более неблагоприятное течение заболевания.
Недуг проявляется в слабости мышц, вплоть до паралича. В большинстве случаев в процесс вовлекаются и дыхательные мышцы. Заболевание развивается вследствие генетического дефекта, приводящего к атрофии определенного рода нервных клеток передних рогов спинного мозга. Встречаемость заболевания составляет от 1:6000 до 1:11000. Диагноз ставится обычно в младенческом или в раннем дошкольном возрасте. Чем раньше появляются первые симптомы, тем более неблагоприятное течение заболевания.
Что такое лечение Spinraza?
Препарат Спинраза впервые позволяет проводить терапию, воздействующую на генетическом уровне, а именно, стимулирующую ген SMN2, который выполняет ту же функцию, что и отсутствующий у больных ген SMN1.
Для обеспечения доставки препарата в нервную систему практикуется интратекальное введение.
Лечение мышечной дистрофии в Медицинском центре Ихилов в Израиле:
В Тель-авивском медицинском центре действует специальная многопрофильная клиника по ведению пациентов с СМА. Руководит ею проф. Паталь, специалист с мировым именем. Пациенты абмулатории — дети и молодые люди.
Руководит ею проф. Паталь, специалист с мировым именем. Пациенты абмулатории — дети и молодые люди.
Первичное освидетельствование производится детским невроло ребенка гом, специализирующимся на нервно-мышечных заболеваниях. Далее пациенты проходят осмотр у специалистов различных профилей:
• детский терапевт
• детский пульмонолог
• детский ортопед
• детский эндокринолог
• детский гастроэнтеролог
• нутриционист
• детский нефролог
• детский кардиолог — по медицинским показаниям.
Мышечная миопатия: симптомы и лечение. Питание и витамины при Миопатии.
Что такое миопатия?
Миопатия – это группа нервно-мышечных заболеваний, которые приводят к постепенной атрофии и дегенерации мышц. Преимущественно это прогрессирующие наследственные патологии. Генетически не обусловленная атрофия мышц также возможна, если человек по той или иной причине (обычно из-за травмы или заболевания, ограничивающего подвижность) не может иметь обычной, повседневной физической нагрузки.
Загадки миопатии
Миопатию принято считать генетическим заболеванием, поскольку более точной информации о причинах ее возникновения у врачей нет. Известно, что при миопатии нарушаются обменные процессы в мышцах и происходит нарушение иннервации – связи мышечной ткани с центральной нервной системой. Так как миопатия зачастую имеет наследственный характер, в основном она проявляется в детском и юношеском возрасте, преимущественно у представителей мужского пола.
Миопатия – это не просто мышечная слабость. Это комплекс нарушений в организме, который приводит к ухудшению деятельности разных систем и органов. Однако наиболее очевидными являются изменения в мышцах, которые приводят к специфическим нарушениям подвижности.
При миопатии нарушается походка, человек ходит с опущенными плечами, что, как правило, вызывает искривление позвоночника (лордоз). Так как заболевание считается прогрессирующим, состояние пациента без соответствующей терапии может значительно ухудшаться. Несколько десятилетий назад миопатия считалась неизлечимой, однако современные медикаментозные средства позволяют замедлить или даже остановить прогрессирование болезни, а также улучшить двигательную активность.
Несколько десятилетий назад миопатия считалась неизлечимой, однако современные медикаментозные средства позволяют замедлить или даже остановить прогрессирование болезни, а также улучшить двигательную активность.
Разные «лица» одной болезни
Существуют формы миопатии, при которых первые симптомы заболевания наблюдаются в детском возрасте, и формы, проявляющиеся только в подростковом или даже юношеском периодах.
Методы лечения миопатии напрямую зависят от ее типа. Особенно это нужно учитывать при назначении физических нагрузок. При миопатии они показаны, однако одним пациентам необходима легкая лечебная физкультура, массаж и нагрузки без переутомления, а другим наоборот – рекомендуют активную гимнастику и энергичные движения.
Роль питания и витаминов при миопатии
При миопатии огромную роль играет правильное питание. Оно должно быть разнообразным и содержать достаточное количество витаминов, микро- и макроэлементов. Важно, чтобы пациент с миопатией получал необходимое количество витаминов группы В – как с продуктами, так и в препаратах.
Кроме того, при миопатии нужно принимать витамин Е, кальций и другие, необходимые для костно-мышечной ткани элементы.
Лечение миопатии проводят невролог, ортопед-травматолог, при необходимости – кардиолог. Чтобы добиться успеха в лечении, нужно своевременно поставить диагноз. Так как заболевание развивается, в основном, в детском и юношеском возрасте, ответственность за своевременное обращение к врачу и адекватное лечение лежит, конечно же, на родителях.
Что делать при атрофии мышц после травм?
В отличие от миопатии, мышечная атрофия, вызванная временной неподвижностью, не прогрессирует. Работа мышц восстанавливается если не в полном, то в значительном объеме (в зависимости от возраста и физического состояния человека), однако для этого приходится приложить определенные усилия.
Для восстановления мышц после вынужденного ограничения движений назначаются умеренные физические нагрузки, часто с использованием тренажеров. Хороший эффект дают массажи, лечебная физкультура, плаванье. Важную роль играет и обеспечение организма питательными веществами, необходимыми для восстановления мышечной активности.
Атрофия – Справочник заболеваний | МЦОЗ
Патологическое состояние, которое сопровождается уменьшением размера, объема и веса целого органа или его отдельных участков с постепенным прекращением функционирования, называется атрофией.
Это заболевание может поражать различные органы и ткани, в частности, мышцы, головной мозг, конечности, сетчатку глаза, кожу. Атрофия может развиться в результате заболеваний, а также как часть естественного процесса старения человека. В связи с этим выделяют старческую и патологическую атрофию.
Атрофия может развиться в результате заболеваний, а также как часть естественного процесса старения человека. В связи с этим выделяют старческую и патологическую атрофию.
Симптомы
В зависимости от характера поражения, локализации, степени выраженности и распространенности, проявляются различные симптомы заболевания. Так, для общей мышечной атрофии характерна худоба, потеря мышечной массы, изможденность. Прогрессирование этой патологии приводит к атрофии клеток мозга и внутренних органов.
При атрофии сетчатки глаза наблюдается потеря четкости зрения, а также возможности различать цвета. По мере ухудшения зрения у больного появляются оптические иллюзии и развивается полная слепота. Атрофия кожи характеризуется потерей эластичности, истончением и сухостью.
Причины
Атрофия — это приобретенный процесс, при котором происходит усыхание тканей и органов.
Для общей атрофии провоцирующими факторами являются:- онкологические заболевания;
- недостаток питательных веществ;
- поражения гипоталамуса;
- инфекционные заболевания, которые протекают на протяжении длительного времени;
- эндокринные нарушения.

- радиационное облучение;
- давление на орган или его часть;
- нагрузки на мышцы, ограничение двигательной активности;
- тяжелая интоксикация организма на фоне серьезных инфекций;
- продолжительный прием гормональных препаратов;
- иннервация;
- нарушение кровообращения вследствие ишемических поражений артерий и вен;
- наследственность;
- дисгормональные нарушения.
Диагностика
В каждом конкретном случае диагностические мероприятия различаются. На начальном этапе при любом типе атрофии лечащий врач назначает физикальное обследование, включающее сбор анамнеза, пальпацию, визуальный осмотр и прочие процедуры. Во всех случаях необходимо проведение лабораторного исследования. Последующая диагностика отличается. Например, для выявления атрофии органа выполняют УЗ-диагностику, МРТ или КТ, рентгенографию, сцинтиграфию, фиброгастродуоденоскопию и прочие процедуры. Основной диагностикой атрофии мышц является биопсия и электромиография. Лабораторная диагностика состоит из оценки определенных показателей в биохимическом и общем анализе крови.
Основной диагностикой атрофии мышц является биопсия и электромиография. Лабораторная диагностика состоит из оценки определенных показателей в биохимическом и общем анализе крови.
Лечение
Начинать лечение необходимо с устранения основного заболевания, которое спровоцировало появление атрофического процесса. В случае, если склеротические поражения и атрофия не сильно запущены, возможно полностью или частично восстановить функции и структуры пораженного органа или его части. Однако глубокие атрофические поражения не поддаются лечению и коррекции.
На особенности лечения влияют тяжесть, форма, длительность заболевания, а также возраст пациента и индивидуальная переносимость медицинских препаратов. В каждом случае врач подбирает методы лечения индивидуально. Как правило, назначается длительное медикаментозное, физиотерапевтическое и симптоматическое лечение. Курс лечения не должен прерываться, и его необходимо регулярно повторять с учетом рекомендаций лечащего врача.
Профилактика
Для профилактики важно своевременное лечение внутренних заболеваний, провоцирующих развитие атрофического процесса.
Лечение спинальной мышечной атрофии в Китае
Лечение спинальной мышечной атрофии в КитаеЛечение спинальной мышечной атрофии
Спинальная мышечная атрофия является аутосомно-рецессивным, генетическим, неврологическим заболеванием. Эта болезнь поражает области нервной системы, которые отвечают за контроль движения скелетных мышц. Клинические проявления характеризуются атрофией мышц, слабостью мышечной ткани, уменьшением общей массы тела, а также отсутствием рефлексов в сухожилиях.
В оздоровительном центре «Илин» специалисты придерживаются методик и традиций медицины Китая. Основное учение китайской медицины — принятие организма человека как единого целого. На этом и построена вся философия и методики китайской медицины. В оздоровительном центре «Илин» используют как базовую и медикаментозную программы лечения, так и альтернативные методы. Также применяется медикаментозная терапия западной медицины.
Основные методы лечения спинальной мышечной атрофии в китайской медицине
Массаж. Проводится ежедневно, курсом 30 дней. Восстанавливает функцию конечностей, способствует насыщению крови кислородом, улучшает подвижность суставов.
Проводится ежедневно, курсом 30 дней. Восстанавливает функцию конечностей, способствует насыщению крови кислородом, улучшает подвижность суставов.
Общая акупунктура. В особые точки на теле вводятся специальные иглы. Длительность курса — 15 дней, через день. Иглоукалывание способствует облегчению болей, улучшению симптомов периферического паралича, а также уменьшению слабости в конечностях.
Восковая терапия. Курс — 15 дней, через день. Глубокое прогревание тканей происходит в результате сжимания и разжимания тканей. Благодаря этому лекарственные препараты быстрее проникают в кровь.
Инъекции. Облегчают симптомы пациентам с дистрофией мышц, а также стимулируют нервную мышечную проводимость. Курс — 15 дней, через день.
Гальванизация. Проводится курсом 15 дней, через день. Способствует улучшению циркуляции крови, стимуляции нервных окончаний, повышению жизнеспособности клеток, стимулированию регенерации нервных волокон, ускорению метаболизма, а также уменьшению мышечного напряжения.
Инъекция лекарственными препаратами. Витамины В1 и В12 способствуют улучшению синергической активности нервов. Стимулируется нервная мышечная проводимость, кровь насыщается кислородом, укрепляются сосуды, происходит питание нервных клеток. Инъекции проводят курсом 10 дней, один раз в три дня.
Медикаментозные методы лечения спинальной мышечной атрофии в китайской традиционной медицине
Особые китайские препараты. Лечат атрофию и слабость мышц.
Капсулы. Назначаются для лечения печени, почек, селезенки, суставов. Снижают утомляемость и повышают иммунитет.
Особые китайские препараты. Принимают для лечения дисбаланса мышечного тонуса, снятия напряжения в мышцах. Способствуют нормализации массы тела и укреплению силы в конечностях.
Суп. Способствует улучшению функционирования желудка, селезенки и почек.
Данные препараты принимаются строго по назначению врача.
Медикаментозное лечение спинальной мышечной атрофии препаратами западной медицины
Специальные инъекции. Назначение: мышечная слабость, нарушение речи, атрофия мышц.
Назначение: мышечная слабость, нарушение речи, атрофия мышц.
Комплекс витаминов В группы. Назначение: восстановление поврежденных нервных тканей, улучшение синаптической передачи нейротрансмиттеров и проводимости нервных волокон.
Инъекции гликозида. Назначают для восстановления поврежденных клеток, стимулирования регенерации нервных волокон, энергосбережения всей системы головного мозга, нейропротекции, а также для улучшения функций переферической и центральной нервных систем.
Внутримышечные нуклеотиды и аминокислоты. Действие: снабжают организм мульти-пептидами, аминокислотами и нуклеотидами, укрепляют нервную систему, улучшают кровообращение, повышают эластичность кровеносных сосудов. Также обеспечивают профилактику атеросклероза и влияют на работу всей нервной системы.
Внутримышечные инъекции фактора роста нервов мыши. Действие: стимулируют регенерацию поврежденных нервных волокон.
Все препараты назначают курсом 15 дней, через день.
Реабилитационные мероприятия:
Трудотерапия. Благодаря использованию различных средств улучшаются координация движения и гибкость тела. Проводится эта процедура ежедневно, в течение 30 дней.
Благодаря использованию различных средств улучшаются координация движения и гибкость тела. Проводится эта процедура ежедневно, в течение 30 дней.
Спортивная терапия. Физическую реабилитацию проводят согласно методам Войта-терапии и Бобат-терапии. Терапия проводится раз в день, длительность — 30 минут. Курс — 30 дней.
Баланс. Физические упражнения помогают тренировать координацию движения и баланса. Пациент выполняет упражнения сидя, лежа на спине, стоя и в движении. Курс — 30 дней, ежедневно по 20 минут.
Альтернативные методы лечения
спинальной мышечной атрофииНаряду с традиционными методами лечения специалисты «Илин» предлагают инновационные методы. Один из них — переливание внутривенно стволовых клеток, которые получены из пуповины новорожденных. Этот метод относительно новый и направлен на улучшение симптомов больных мышечной дистонией. Цена 4200 $.
Второй метод — введение эмбриональных стволовых клеток. Процедура улучшает умственные способности и координацию движения. Проводится 4 раза в неделю, длительность процедуры — 45 минут. Стоимость этого курса 4560$.
Проводится 4 раза в неделю, длительность процедуры — 45 минут. Стоимость этого курса 4560$.
Цена лечения спинальной мышечной атрофии в Китае
Точная стоимость курса реабилитации рассчитывается, когда пациент приезжает в клинику. Приблизительная стоимость курса на месяц — 6700$. Каждому пациенту назначается комплексное индивидуальное лечение. Именно оно влияет на окончательную общую стоимость. В базовую оплату входят: базовая программа лечения, обследование и трансфер в обе стороны. Дополнительные расходы: питание, проживание, оформление визы и авиабилеты. За дополнительную плату также назначаются альтернативные методы лечения, но только после консультации у лечащего врача. Проживание пациентов предусматривается в стандартных номерах больницы «Илин» или оздоровительного центра. Цена за номер: 30$ или 70$ соответственно. В больнице «Илин» также расположены VIP номера по цене 150$.
Полиомиелит это – атрофия мышц, деформация конечностей и туловища, инвалидизация
ПРЕСС–РЕЛИЗ
24 октября 2019 года
Всемирный день борьбы с полиомиелитом
Государственное учреждение «Центр общественного здоровья Министерства здравоохранения Украины» сообщает, что 24. 10.2019 года в Украине будет отмечаться «Всемирный День борьбы с полиомиелитом». Он проводится в связи с необходимостью информирования населения страны об опасности полиомиелита и мероприятиях, проводимых на государственном и административном уровнях по профилактике и эпиднадзора за этой болезнью.
10.2019 года в Украине будет отмечаться «Всемирный День борьбы с полиомиелитом». Он проводится в связи с необходимостью информирования населения страны об опасности полиомиелита и мероприятиях, проводимых на государственном и административном уровнях по профилактике и эпиднадзора за этой болезнью.
Статистика по Украине и Харьковской области
«Харьковский областной лабораторный центр МЗ Украины» сообщает, что по данным «Центра общественного здоровья МОЗ Украины», в Украине за период с января по 15 сентября 2019 зарегистрировано 99 случаев острых вялых параличей (ОВС). Выявлено 29 «горячих» случаев ОВП, в частности, по причине отсутствия 3-х прививок против полиомиелита у заболевших.
Из обследованных объектов окружающей среды наибольшее количество вирусов 59 (2,0%) было выделено из сточной воды, среди которых 28 (0,9%) различные штаммы полиовирусов.
В целом по Украине показатели неудовлетворительные: Полио-3 (дети до года) — 45,5%; Полио-4 (18 мес.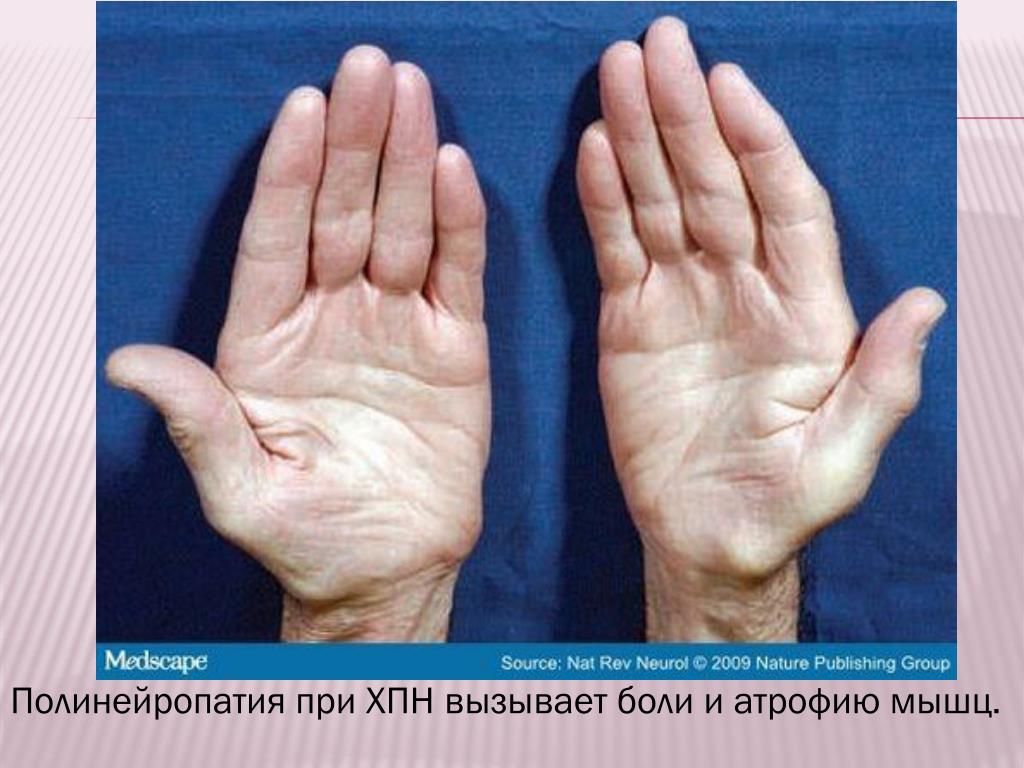 ) — 45,2%; Полио-5 (6 лет) — 52,3%; Полио-6 (14 лет) — 54,0%. Особое беспокойство вызывают показатели охвата прививками детей раннего возраста, которые не достигают контрольного уровня: за 7 мес. — 58,1%. Это свидетельствует о недостаточной реализацию мероприятий иммунопрофилактики по поддержке статуса страны, свободной от полиомиелита.
) — 45,2%; Полио-5 (6 лет) — 52,3%; Полио-6 (14 лет) — 54,0%. Особое беспокойство вызывают показатели охвата прививками детей раннего возраста, которые не достигают контрольного уровня: за 7 мес. — 58,1%. Это свидетельствует о недостаточной реализацию мероприятий иммунопрофилактики по поддержке статуса страны, свободной от полиомиелита.
По данным отдела мониторинга иммунопрофилактики и защиты населения от инфекционных болезней КНП ХОР «Областная детская инфекционная клиническая больница» за 8 месяцев 2019 объем профилактических прививок против полиомиелита выполнено следующим образом: Полио-3 (до года) — 42,2%, Полио-3 (1 год и старше) — 40,5%, Полио-4 (18 мес.) — 43,7%, Полио-4 (ст. 2 лет) — 54,8%, Полио-5 (6 лет) — 62,9%, Полио-5 (ст. 7 лет) — 69,8%, Полио-6 (14 лет) — 60,9%, Полио-6 (ст.15 лет) — 45,0%.
Что обязательно надо знать о полиомиелите
Полиомиелит (детский спинальный паралич) – острое инфекционное заболевание, поражающее ткани спинного мозга.
Возбудителем болезни является полиовирус. Инфекция передается от больного человека фекально-оральным путем, реже – воздушно-капельным. Чаще всего полиомиелитом болеют дети в возрасте до 10 лет. В наше время периодически регистрируются отдельные случаи заболевания поломиелитом. Они связаны с несоблюдением регламентированных сроков вакцинации, а также с отказом некоторых родителей от прививок их детей против полиомиелита. В довакцинальный период паралитический полиомиелит поражал тысячи и десятки тысяч детей, из которых 40% оставались инвалидами.
Заболевание начинается с резкого повышения температуры тела. В первые дни появляются симптомы, характерные для острой респираторной инфекции (насморк, боль в горле, сухой кашель), иногда к ним присоединяются признаки расстройства пищеварения (тошнота, рвота, боль в животе). Эти симптомы могут исчезать на несколько дней, после чего состояние больного ухудшается – усиливается головная боль, возникает боль в мышцах, повышается чувствительность кожи к любым раздражителям.
При паралитической форме полиомиелита из-за поражения двигательных нейронов спинного мозга наступает нарушение функции мышц, которая проявляется парезами и параличами. Активное движение конечностями становится ограниченным или вовсе невозможным. Иногда могут развиваться параличи мышц шеи, туловища.
Паралитическая стадия болезни продолжается от нескольких дней до 2 недель. Восстановление мышечных функций может длиться месяцами и даже годами. При глубоком поражении двигательных нейронов спинного мозга, процесс восстановления становится невозможным и после перенесенной болезни остаются на всю жизнь тяжелые ее последствия: атрофия мышц, деформация конечностей и туловища.
Надёжной защитой от этой опасной инфекции является только вакцинация.
Профилактические прививки против полиомиелита введены в национальный календарь обязательной иммунизации и проводятся детям с двухмесячного возраста.
Справочная информация о дате
Всемирный день борьбы с полиомиелитом был учрежден Ротари Интернэшнл более 10 лет назад. Его дата – 24 октября – приурочена ко дню рождения Джонаса Солка, под чьим руководством была создана первая успешная вакцина против полиомиелита. Эффективное использование этой инактивированной полиовирусной вакцины и, в дальнейшем, широкое распространение оральной полиомиелитной вакцины, созданной Альбертом Сэйбином, позволило Всемирной ассамблее здравоохранения принять в 1998 г. резолюцию по вопросу искоренения полиовируса. С тех пор ВОЗ, Ротари Интернэшнл и другие партнеры по Глобальной инициативе по ликвидации полиомиелита интенсивно работали над снижением бремени этой болезни во всем мире, и в 2016 г. было зарегистрировано рекордно низкое число случаев полиомиелита. Искоренить полиомиелит реально, и это станет одним из величайших достижений за всю историю общественного здравоохранения, которое позволит защитить от болезни всех детей – и живущих сейчас, и тех, которые родятся в будущем.
Его дата – 24 октября – приурочена ко дню рождения Джонаса Солка, под чьим руководством была создана первая успешная вакцина против полиомиелита. Эффективное использование этой инактивированной полиовирусной вакцины и, в дальнейшем, широкое распространение оральной полиомиелитной вакцины, созданной Альбертом Сэйбином, позволило Всемирной ассамблее здравоохранения принять в 1998 г. резолюцию по вопросу искоренения полиовируса. С тех пор ВОЗ, Ротари Интернэшнл и другие партнеры по Глобальной инициативе по ликвидации полиомиелита интенсивно работали над снижением бремени этой болезни во всем мире, и в 2016 г. было зарегистрировано рекордно низкое число случаев полиомиелита. Искоренить полиомиелит реально, и это станет одним из величайших достижений за всю историю общественного здравоохранения, которое позволит защитить от болезни всех детей – и живущих сейчас, и тех, которые родятся в будущем.
Уважаемые родители!
Не отказывайтесь от прививок, подумайте
о последствиях для своих детей
Прогрессивная мышечная атрофия — это.
 .. Что такое Прогрессивная мышечная атрофия?
.. Что такое Прогрессивная мышечная атрофия?хроническая болезнь, существенный симптом которой заключается в постепенно возрастающем и распространяющемся исчезании мышечных волокон. По мере этого исчезания падает сократительная способность мышцы, и от этого все более затрудняется и наконец совершенно исчезает возможность передвижения членов. Болезнь эта поражает только поперечно-полосатые мышцы скелета, необходимые для произвольных движений, и оставляет нетронутыми гладкие мышцы внутренностей, а также сердечную мышцу. Поэтому растительные отправления, совершающиеся с участием последних, протекают более или менее правильно и жизнь может продолжаться неопределенно долгое время, когда болезненный процесс, лежащий в основе прогрессивной мышечной атрофии, достигает даже крайних размеров. Иногда доходит до того, что больной не может шевельнуть ни одним пальцем, не может изменить положения своего тела, руки и ноги висят как плети, голова от тяжести падает взад или вперед и т. п. Исхудание конечностей и туловища вследствие исчезания мышц доходит до такой степени, что человек в буквальном смысле представляет из себя лишь кожу да кости. В большинстве случаев, впрочем, наступает смерть от какого-нибудь осложнения раньше, чем процесс успеет дойти до крайней степени. Обычно болезнь развивается весьма медленно и длится многие годы. Существенные жизненные отправления, как то: кровообращение, дыхание, пищеварение, глотание, а также специальные функции нервной системы, остаются свободными от поражения. В целом ряде случаев, составляющих классическую форму болезни, она прежде всего поражает мелкие мышцы ручной кисти, затем мускулатуру плеча, а впоследствии мышцы нижних конечностей и туловища. В других, также довольно частых случаях дело начинается с атрофии мышц плечевого и тазового пояса и лишь спустя долгое время распространяется на периферические части конечностей. Встречаются и другие сочетания, в том числе и такие, при которых болезнь первоначально поражает мышцы лица. Замечательно, что иногда болезнь эта наблюдается у нескольких членов одной и той же семьи, причем она у всех протекает по одинаковому типу. В известном ряде случаев в основе болезни лежит хроническое заболевание тех отделов спинного мозга, которые заключают в себе трофические центры для мышц.
В большинстве случаев, впрочем, наступает смерть от какого-нибудь осложнения раньше, чем процесс успеет дойти до крайней степени. Обычно болезнь развивается весьма медленно и длится многие годы. Существенные жизненные отправления, как то: кровообращение, дыхание, пищеварение, глотание, а также специальные функции нервной системы, остаются свободными от поражения. В целом ряде случаев, составляющих классическую форму болезни, она прежде всего поражает мелкие мышцы ручной кисти, затем мускулатуру плеча, а впоследствии мышцы нижних конечностей и туловища. В других, также довольно частых случаях дело начинается с атрофии мышц плечевого и тазового пояса и лишь спустя долгое время распространяется на периферические части конечностей. Встречаются и другие сочетания, в том числе и такие, при которых болезнь первоначально поражает мышцы лица. Замечательно, что иногда болезнь эта наблюдается у нескольких членов одной и той же семьи, причем она у всех протекает по одинаковому типу. В известном ряде случаев в основе болезни лежит хроническое заболевание тех отделов спинного мозга, которые заключают в себе трофические центры для мышц. Кроме того, и заболевание периферических нервов может привести к П. мышечной атрофии. Таким образом, эта болезнь иногда представляет результат центрального или периферического страдания нервной системы, иногда же самостоятельное страдание мышечной системы организма. Что касается анатомических изменений в пораженных мышцах, то они сводятся на исчезание своеобразной ткани, обусловливающей сократительность мышц, и на замену ее соединительной тканью. Нередко местами вместо последней развивается в обильном количестве жировая ткань, и тогда соответственное место на руке или ноге представляет вместо исхудания утолщение, как будто мышечная масса здесь увеличилась. Эта кажущаяся прибыль мышц называется ложной гипертрофией (псевдогипертрофия мышц).
Кроме того, и заболевание периферических нервов может привести к П. мышечной атрофии. Таким образом, эта болезнь иногда представляет результат центрального или периферического страдания нервной системы, иногда же самостоятельное страдание мышечной системы организма. Что касается анатомических изменений в пораженных мышцах, то они сводятся на исчезание своеобразной ткани, обусловливающей сократительность мышц, и на замену ее соединительной тканью. Нередко местами вместо последней развивается в обильном количестве жировая ткань, и тогда соответственное место на руке или ноге представляет вместо исхудания утолщение, как будто мышечная масса здесь увеличилась. Эта кажущаяся прибыль мышц называется ложной гипертрофией (псевдогипертрофия мышц).
Какие болезнетворные причины обусловливают развитие этого тяжкого страдания, совершенно неизвестно. По-видимому, чрезмерное напряжение мышц усиленной работой подготавливает для него почву. По всей вероятности, главное условие составляет врожденное предрасположение. Лечение П. мышечной атрофии до настоящего времени крайне безуспешно: преимущественно применяется электричество.
Лечение П. мышечной атрофии до настоящего времени крайне безуспешно: преимущественно применяется электричество.
П. Розенбах.
Спинальная мышечная атрофия (СМА): типы, симптомы и лечение
Обзор
Что такое спинальная мышечная атрофия (СМА)?
Спинальная мышечная атрофия (СМА) — это генетическое (наследственное) нервно-мышечное заболевание, при котором мышцы становятся слабыми и истощаются. Люди с СМА теряют нервные клетки определенного типа в спинном мозге (называемые мотонейронами), которые контролируют движение мышц.Без этих мотонейронов мышцы не получают нервных сигналов, которые заставляют мышцы двигаться. Слово атрофия — это медицинский термин, который означает меньше. При СМА некоторые мышцы становятся меньше и слабее из-за недостаточной активности.
Насколько распространена мышечная атрофия позвоночника?
Приблизительно от 10 000 до 25 000 детей и взрослых живут с СМА в Соединенных Штатах. Это редкое заболевание, которым страдает один из 6000–10 000 детей.
Это редкое заболевание, которым страдает один из 6000–10 000 детей.
У кого может развиться мышечная атрофия позвоночника?
Человек с СМА наследует две копии отсутствующего или дефектного (мутировавшего) гена моторного нейрона выживания 1 (SMN1).Один дефектный ген происходит от матери, а другой — от отца. Взрослый человек может иметь единственную копию дефектного гена, вызывающего СМА, и не знать об этом.
Около шести миллионов американцев (1 из 50) являются носителями мутированного гена SMN1. Эти носители имеют один здоровый ген SMN1 и один отсутствующий или дефектный ген SMN1. У перевозчиков не развивается SMA. Вероятность того, что у двух носителей будет ребенок с СМА, составляет 1 из 4.
Какие виды спинномозговой мышечной атрофии?
Существует четыре основных типа SMA:
- Тип 1 (тяжелый): Около 60% людей со СМА имеют тип 1, также называемый болезнью Верднига-Хоффмана.Симптомы появляются при рождении или в течение первых шести месяцев жизни младенца. Младенцы с СМА типа 1 испытывают трудности с глотанием и сосанием. Они не справляются с обычными вехами, такими как поднятие головы или сидение. По мере того как мышцы продолжают ослабевать, дети становятся более склонными к респираторным инфекциям и коллапсу легких (пневмотораксу). Большинство детей с СМА типа 1 умирают до своего второго дня рождения.
- Тип 2 (промежуточный): Симптомы СМА 2 типа (также называемой болезнью Дубовица) появляются, когда ребенку от шести до 18 месяцев.Этот тип чаще поражает нижние конечности. Дети со СМА 2 типа могут сидеть, но не могут ходить. Большинство детей со СМА 2 типа доживают до взрослого возраста.
- Тип 3 (легкая форма): Симптомы СМА 3 типа (также называемая СМА Кугельберта-Веландера или СМА с ювенильным началом) проявляются после первых 18 месяцев жизни ребенка. У некоторых людей с типом 3 симптомы болезни проявляются только в раннем взрослом возрасте. Симптомы 3-го типа включают легкую мышечную слабость, затруднения при ходьбе и частые респираторные инфекции. Со временем симптомы могут повлиять на способность ходить или стоять. СМА типа 3 не приводит к значительному сокращению продолжительности жизни.
- Тип 4 (взрослый): Редкая форма СМА у взрослых обычно не появляется до середины 30 лет. Симптомы мышечной слабости прогрессируют медленно, поэтому большинство людей с типом 4 остаются подвижными и живут полноценной жизнью.
 Младенцы с СМА типа 1 испытывают трудности с глотанием и сосанием. Они не справляются с обычными вехами, такими как поднятие головы или сидение. По мере того как мышцы продолжают ослабевать, дети становятся более склонными к респираторным инфекциям и коллапсу легких (пневмотораксу). Большинство детей с СМА типа 1 умирают до своего второго дня рождения.
Младенцы с СМА типа 1 испытывают трудности с глотанием и сосанием. Они не справляются с обычными вехами, такими как поднятие головы или сидение. По мере того как мышцы продолжают ослабевать, дети становятся более склонными к респираторным инфекциям и коллапсу легких (пневмотораксу). Большинство детей с СМА типа 1 умирают до своего второго дня рождения. Со временем симптомы могут повлиять на способность ходить или стоять. СМА типа 3 не приводит к значительному сокращению продолжительности жизни.
Со временем симптомы могут повлиять на способность ходить или стоять. СМА типа 3 не приводит к значительному сокращению продолжительности жизни.Симптомы и причины
Что вызывает спинальную мышечную атрофию?
У людей с СМА отсутствует часть гена SMN1 или изменен (мутирован) ген.Здоровый ген SMN1 производит белок SMN. Моторным нейронам нужен этот белок, чтобы выжить и нормально функционировать.
Люди с СМА не вырабатывают достаточное количество белка SMN, поэтому мотонейроны сжимаются и умирают. В результате мозг не может контролировать произвольные движения, особенно движения в голове, шее, руках и ногах.
У людей также есть гены SMN2, которые производят небольшое количество белка SMN. У человека может быть до восьми копий гена SMN2. Наличие нескольких копий гена SMN2 обычно приводит к менее серьезным симптомам СМА, поскольку дополнительные гены восполняют недостающий белок SMN1.В редких случаях СМА вызывают мутации гена, не относящегося к SMN (не хромосомы 5).
Каковы симптомы спинальной мышечной атрофии?
СимптомыСМА различаются в зависимости от типа. В целом, люди с СМА испытывают прогрессирующую потерю мышечного контроля, движения и силы. Потеря мышечной массы ухудшается с возрастом. Заболевание имеет тенденцию серьезно поражать мышцы, расположенные ближе всего к туловищу и шее. Некоторые люди с СМА никогда не ходят, не сидят и не стоят. Другие постепенно теряют способность выполнять эти действия.
Диагностика и тесты
Как диагностируется мышечная атрофия позвоночника?
Некоторые симптомы СМА напоминают симптомы нервно-мышечных расстройств, таких как мышечная дистрофия. Чтобы определить причину симптомов, ваш лечащий врач проведет медицинский осмотр и получит историю болезни. Ваш врач может также назначить один или несколько из этих тестов для диагностики SMA:
Чтобы определить причину симптомов, ваш лечащий врач проведет медицинский осмотр и получит историю болезни. Ваш врач может также назначить один или несколько из этих тестов для диагностики SMA:
- Анализ крови: Анализ крови на ферменты и белок позволяет проверить высокий уровень креатинкиназы. Ухудшенные мышцы высвобождают этот фермент в кровоток.
- Генетический тест: Этот анализ крови выявляет проблемы с геном SMN1. В качестве диагностического инструмента генетический тест на 95% эффективен при обнаружении измененного гена SMN1.В некоторых штатах тест на СМА проводится в рамках плановых обследований новорожденных.
- Тест нервной проводимости: Электромиограмма (ЭМГ) измеряет электрическую активность нервов, мышц и нервов.
- Биопсия мышцы: В редких случаях врач может выполнить биопсию мышцы. Эта процедура включает удаление небольшого количества мышечной ткани и отправку ее в лабораторию для исследования. Биопсия может показать атрофию или потерю мышечной массы.
 Биопсия может показать атрофию или потерю мышечной массы.
Биопсия может показать атрофию или потерю мышечной массы.Можно ли диагностировать мышечную атрофию позвоночника во время беременности?
Если вы беременны и имеете семейный анамнез СМА, пренатальные тесты могут определить, есть ли у вашего будущего ребенка заболевание.Эти тесты немного повышают риск выкидыша или прерывания беременности. Пренатальные тесты на SMA включают:
- Амниоцентез: Во время амниоцентеза ваш акушер вводит вам в живот тонкую иглу, чтобы извлечь небольшое количество жидкости из амниотического мешка. Лабораторный специалист (патолог) проверяет жидкость на наличие СМА. Этот тест проводится после 14 недели беременности.
- Взятие пробы ворсинок хориона (CVS): Ваш акушер берет небольшой образец ткани из плаценты через шейку матки или желудок.Патологоанатом проверяет образец на SMA. CVS может иметь место уже на 10 неделе беременности.
Ведение и лечение
Как лечить или лечить спинальную мышечную атрофию?
Нет лекарства от SMA. Лечение зависит от типа и симптомов СМА.Многие люди с СМА получают пользу от физиотерапии и трудотерапии и вспомогательных устройств, таких как ортопедические скобы, костыли, ходунки и инвалидные коляски.
Лечение зависит от типа и симптомов СМА.Многие люди с СМА получают пользу от физиотерапии и трудотерапии и вспомогательных устройств, таких как ортопедические скобы, костыли, ходунки и инвалидные коляски.
Эти процедуры также могут помочь:
- Терапия, изменяющая заболевание: Эти препараты стимулируют выработку белка SMN. Нусинерсен (Спинраза®) предназначен для детей в возрасте от 2 до 12 лет. Ваш врач вводит препарат в пространство вокруг позвоночного канала. Другой препарат, рисдаплам (Эврисди®), помогает взрослым и детям старше двух месяцев.Люди принимают рисдаплам ежедневно внутрь (перорально).
- Заместительная генная терапия: Детям младше двух лет может помочь однократная внутривенная (IV) инфузия препарата, называемого онасемногеном абепарвовец-xioi (Zolgensma®). Эта терапия заменяет отсутствующий или неисправный ген SMN1 функционирующим геном.
Каковы осложнения мышечной атрофии позвоночника?
Со временем люди с СМА испытывают прогрессирующую мышечную слабость и потерю мышечного контроля. Возможные осложнения включают:
Возможные осложнения включают:
Профилактика
Как предотвратить мышечную атрофию позвоночника?
СМА — наследственное заболевание. Если вы или ваш партнер несете мутировавший ген, вызывающий СМА, генетический консультант может объяснить шансы, что ваш ребенок болен СМА или является носителем.
Возможно, вы сможете принять меры до беременности, чтобы снизить риск передачи СМА. Процесс, называемый предимплантационной генетической диагностикой (ПГД), позволяет идентифицировать эмбрионы, у которых нет мутировавшего гена. Ваш врач имплантирует здоровые эмбрионы во время экстракорпорального оплодотворения (ЭКО). PGD гарантирует, что ваш ребенок будет иметь два здоровых гена SMN1 и не заболеет SMA.
Перспективы / Прогноз
Каков прогноз (перспективы) для людей со спинальной мышечной атрофией?
Качество и продолжительность жизни людей с СМА варьируются в зависимости от типа. Младенцы со СМА типа 1 обычно умирают до своего второго дня рождения. Дети с СМА 2 или 3 типа могут жить полноценной жизнью в зависимости от тяжести симптомов. Люди, у которых СМА развивается в зрелом возрасте (тип 4), часто остаются активными и имеют нормальную продолжительность жизни.
Младенцы со СМА типа 1 обычно умирают до своего второго дня рождения. Дети с СМА 2 или 3 типа могут жить полноценной жизнью в зависимости от тяжести симптомов. Люди, у которых СМА развивается в зрелом возрасте (тип 4), часто остаются активными и имеют нормальную продолжительность жизни.
Жить с
Когда мне позвонить врачу?
Вы должны позвонить своему врачу, если у кого-то есть SMA:
Какие вопросы я должен задать своему врачу?
Вы можете спросить:
- Как я или мой ребенок заболели СМА?
- Какой тип SMA у меня или моего ребенка?
- Каков прогноз для этого типа SMA?
- Как лучше всего лечить этот тип СМА?
- Каковы риски лечения и побочные эффекты?
- Подвержены ли другие члены семьи риску заболеть СМА? Если да, должны ли мы сдавать генетические тесты?
- Какой вид постоянного ухода понадобится мне или моему ребенку?
- Стоит ли обращать внимание на признаки осложнений?
Записка из клиники Кливленда
SMA — это генетическое нервно-мышечное заболевание, которое может существенно повлиять на качество и продолжительность жизни. Это прогрессирующее заболевание, которое со временем ухудшается. Симптомы могут присутствовать при рождении (тип 1), развиваться в детстве (тип 2 или 3) или во взрослом возрасте (тип 4). Многообещающие новые методы лечения заболеваний и замены генов. Можно нести ген, вызывающий СМА, и не знать об этом. Если СМА присутствует в вашей семье, поговорите со своим врачом о том, как снизить вероятность развития СМА у вашего будущего ребенка.
Это прогрессирующее заболевание, которое со временем ухудшается. Симптомы могут присутствовать при рождении (тип 1), развиваться в детстве (тип 2 или 3) или во взрослом возрасте (тип 4). Многообещающие новые методы лечения заболеваний и замены генов. Можно нести ген, вызывающий СМА, и не знать об этом. Если СМА присутствует в вашей семье, поговорите со своим врачом о том, как снизить вероятность развития СМА у вашего будущего ребенка.
Атрофия мышц — обзор
22.2.3 Атрофия
Атрофия мышц может быть вызвана как физиологическими, так и болезненными состояниями. Исследования показали, что активация E3 ubiquitin ligases MuRF1 и MAFbx играет важную роль в мышечной атрофии (Bodine et al., 2001; Gomes et al., 2001). Убиквитинлигазы Е3 являются важной частью протеасомного пути, который отвечает за деградацию белка в клетках. В клетках белки, предназначенные для деградации протеасом, маркируются убиквитинлигазами. Выбор субстрата для деградации белка зависит от убиквитинлигазы E3. Было показано, что MuRF1 и MAFbx нацелены на специфические для мышц белки для деградации в различных условиях, включая неиспользование мышц, иммобилизацию, денервацию и лечение стероидами (McElhinny et al., 2002; Clarke et al., 2007; Tintignac et al., 2005; Lagirand -Cantaloube et al., 2008). MuRF1 и MAFbx регулируются факторами транскрипции FOXO (Sandri et al., 2004, 2006; Stitt et al., 2004). Активность FOXO регулируется сигнализацией инсулина и факторов роста. Исследования показали, что инсулин и IGF активируют передачу сигналов PI3K / AKT, что приводит к фосфорилированию FOXO (Biggs et al., 1999; Брюнет и др., 1999, 2001; Копс и др., 1999). Когда FOXO не фосфорилируется, он локализуется в ядрах и активирует нижестоящие мишени транскрипции, такие как MuRF1, MAFbx и другие гены, которые способствуют атрофии мышц. Когда он фосфорилируется, он исключается из ядра, поэтому транскрипционная активность FOXO подавляется, что снижает экспрессию MuRF1 и MAFbx. Было показано, что помимо FOXO активация передачи сигналов NFκB и p38 MAPK, активируемая воспалительными сигналами и окислительным стрессом, активирует транскрипцию MuRF1 и / или MAFbx, способствуя атрофии мышц (Brunet et al.
Было показано, что MuRF1 и MAFbx нацелены на специфические для мышц белки для деградации в различных условиях, включая неиспользование мышц, иммобилизацию, денервацию и лечение стероидами (McElhinny et al., 2002; Clarke et al., 2007; Tintignac et al., 2005; Lagirand -Cantaloube et al., 2008). MuRF1 и MAFbx регулируются факторами транскрипции FOXO (Sandri et al., 2004, 2006; Stitt et al., 2004). Активность FOXO регулируется сигнализацией инсулина и факторов роста. Исследования показали, что инсулин и IGF активируют передачу сигналов PI3K / AKT, что приводит к фосфорилированию FOXO (Biggs et al., 1999; Брюнет и др., 1999, 2001; Копс и др., 1999). Когда FOXO не фосфорилируется, он локализуется в ядрах и активирует нижестоящие мишени транскрипции, такие как MuRF1, MAFbx и другие гены, которые способствуют атрофии мышц. Когда он фосфорилируется, он исключается из ядра, поэтому транскрипционная активность FOXO подавляется, что снижает экспрессию MuRF1 и MAFbx. Было показано, что помимо FOXO активация передачи сигналов NFκB и p38 MAPK, активируемая воспалительными сигналами и окислительным стрессом, активирует транскрипцию MuRF1 и / или MAFbx, способствуя атрофии мышц (Brunet et al. , 2004; Cai et al., 2004; Мастрокола и др., 2008; Powers et al., 2007).
, 2004; Cai et al., 2004; Мастрокола и др., 2008; Powers et al., 2007).
Сообщается, что помимо протеасомной деградации белков, нерегулируемая активность аутофагии и активация каспазы-3 и кальпаинов также играют важную роль в атрофии скелетных мышц (Salazar et al., 2010; Enns and Belcastro, 2006; Milan et al. ., 2015; Nascimbeni et al., 2012). Аутофагия удаляет поврежденные или нежелательные органеллы и белки в клетках. Процесс включает координацию группы генов, связанных с аутофагией, которые кодируют белки, избирательно взаимодействующие с мишенями, образование аутофагосом, слияние с лизосомами и деградацию целевых органелл и белков.Несбалансированная активность аутофагии в клетках связана с истощением мышц и заболеваниями. В то время как базальный уровень активности аутофагии необходим для поддержания здоровья мышц, чрезмерная аутофагия вызывает истощение мышц. FOXO3 индуцирует экспрессию ряда генов аутофагии, участвующих в различных стадиях процесса, включая LC3b, Gabarapl1, Pi3kIII, Ulk2, Atg12l, Beclin1, Atg4b и Bnip3 (Zhao et al. , 2007; Mammucari et al., 2007) . Кроме того, предыдущие исследования показали, что FOXO активирует аутофагию в дополнение к деградации протеасом, которая включает митофагию, особую форму аутофагии (Milan et al., 2015; Чжао и др., 2008). Аутофагия в вышедших из употребления скелетных мышцах также может быть активирована с помощью передачи сигналов p38 MAPK (McClung et al., 2010).
, 2007; Mammucari et al., 2007) . Кроме того, предыдущие исследования показали, что FOXO активирует аутофагию в дополнение к деградации протеасом, которая включает митофагию, особую форму аутофагии (Milan et al., 2015; Чжао и др., 2008). Аутофагия в вышедших из употребления скелетных мышцах также может быть активирована с помощью передачи сигналов p38 MAPK (McClung et al., 2010).
Увеличение количества некоторых циркулирующих сигнальных молекул, таких как воспалительные цитокины (например, TNFα, IL1β и IL6), TGFβ и стероиды, может активировать молекулярные пути, которые вызывают атрофию мышц (Bruunsgaard and Pedersen, 2003; Schakman et al., 2012; Spate и Schulze, 2004; Watson et al., 2012; Narola et al., 2013). Также было показано, что оксидативный стресс способствует мышечной атрофии.Было показано, что оба активируют передачу сигналов NFκB и / или MAPK, которые снижают дифференцировку миобластов, вызывают апоптоз и увеличивают деградацию белка (Powers et al., 2011; Archuleta et al. , 2009; Langen et al., 2012; Hunter et al. , 2002; Лу и др., 2012). Кроме того, было показано, что IL6 активирует STAT3 и способствует развитию кахексии и саркопении при раке (Budui et al., 2015; Bonetto et al., 2012; Gilabert et al., 2014). Было показано, что помимо воспалительных цитокинов, передача сигналов TGFβ способствует мышечной атрофии (Narola et al., 2013; Mendias et al., 2012). Было показано, что и TGFβ1, и миостатин активируют smad2 / 3 и приводят к истощению мышц. Мы также недавно сообщили о новом взаимодействии между передачей сигналов TGFβ1 и STAT3, которые вносят вклад в более серьезное мышечное истощение в условно-специфической мышечной модели TGFβ1 у мышей (Guadagnin et al., 2015).
, 2009; Langen et al., 2012; Hunter et al. , 2002; Лу и др., 2012). Кроме того, было показано, что IL6 активирует STAT3 и способствует развитию кахексии и саркопении при раке (Budui et al., 2015; Bonetto et al., 2012; Gilabert et al., 2014). Было показано, что помимо воспалительных цитокинов, передача сигналов TGFβ способствует мышечной атрофии (Narola et al., 2013; Mendias et al., 2012). Было показано, что и TGFβ1, и миостатин активируют smad2 / 3 и приводят к истощению мышц. Мы также недавно сообщили о новом взаимодействии между передачей сигналов TGFβ1 и STAT3, которые вносят вклад в более серьезное мышечное истощение в условно-специфической мышечной модели TGFβ1 у мышей (Guadagnin et al., 2015).
Спинальная мышечная атрофия — NHS
Спинальная мышечная атрофия (СМА) — это генетическое заболевание, которое ослабляет мышцы и вызывает проблемы с движением.
Это серьезное заболевание, которое со временем ухудшается, но существуют методы лечения, которые помогают справиться с симптомами.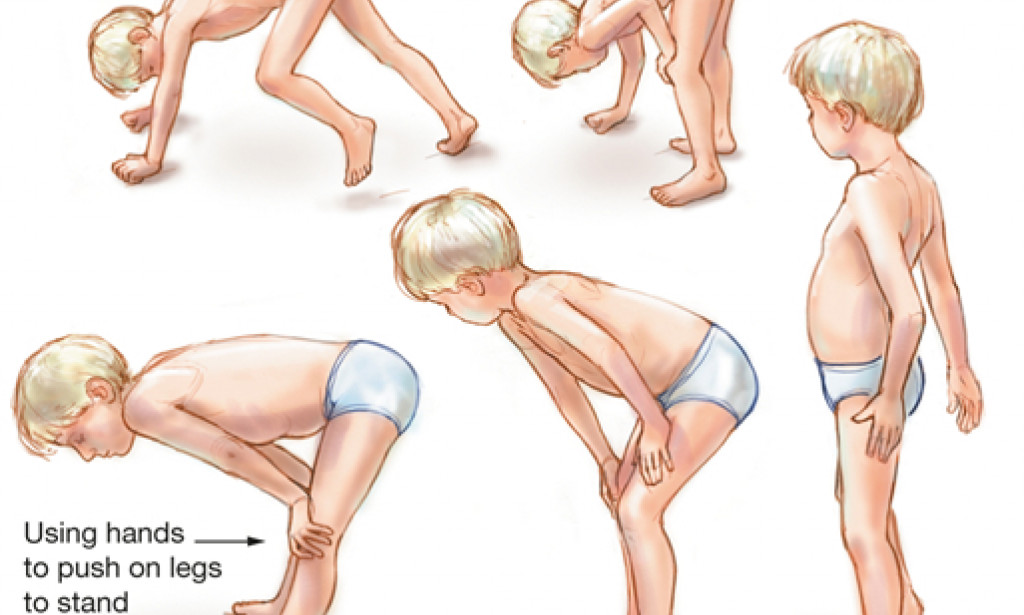
Симптомы SMA
Симптомы SMA и время их появления зависят от типа вашего SMA.
Типичные симптомы включают:
- неустойчивые или слабые руки и ноги
- проблемы с движением, такие как затруднение при сидении, ползании или ходьбе
- подергивание или тряска мышц (тремор)
- проблемы с костями и суставами, такие как необычно изогнутые позвоночник (сколиоз)
- Проблемы с глотанием
- Проблемы с дыханием
СМА не влияет на интеллект и не вызывает трудностей в обучении.
Типы SMA
Существует несколько типов SMA, которые начинаются в разном возрасте. Некоторые типы вызывают более серьезные проблемы, чем другие.
Основными типами являются:
- тип 1 — развивается у детей младше 6 месяцев и является наиболее тяжелым типом
- Тип 2 — появляется у детей в возрасте от 7 до 18 месяцев и менее тяжел, чем тип 1
- тип 3 — развивается после 18 месяцев и является наименее тяжелым типом, поражающим детей
- тип 4 — поражает взрослых и обычно вызывает только легкие проблемы
В прошлом младенцы с типом 1 редко выживали дольше первых нескольких лет жизни. жизнь.Но в последние годы результаты улучшились благодаря ранней диагностике и лечению.
жизнь.Но в последние годы результаты улучшились благодаря ранней диагностике и лечению.
Большинство детей с типом 2 доживают до взрослого возраста и могут прожить долгую полноценную жизнь. Типы 3 и 4 обычно не влияют на продолжительность жизни.
Подробнее о типах SMA.
Лечение SMA
В настоящее время вылечить СМА невозможно, но исследования по поиску возможных новых методов лечения продолжаются.
Доступны лечение и поддержка для лечения симптомов и помощи людям с СМА в достижении наилучшего качества жизни.
Лечение может включать:
- упражнения и оборудование для улучшения движения и дыхания
- трубки для кормления и рекомендации по диете
- скобки или хирургическое вмешательство для лечения проблем с позвоночником или суставами
К участию в лечении могут быть привлечены различные медицинские работники ваше медицинское обслуживание, включая врачей-специалистов, физиотерапевтов, эрготерапевтов, логопедов и лингвистов.
Подробнее о лечении СМА.
Тесты на SMA
Генетическая проблема, вызывающая СМА, передается ребенку от родителей.
Поговорите с терапевтом, если вы планируете беременность и:
- у вас был ребенок с СМА до
- у вас есть история болезни в вашей семье
- у вашего партнера есть история болезни в их семья
Врач общей практики может направить вас к консультанту по генетическим вопросам, чтобы обсудить риск заболевания, влияющего на будущую беременность, и любые возможные СПС.
Если вы беременны и есть вероятность, что у вашего ребенка может быть СМА, можно провести тесты, чтобы проверить, родится ли он с этим заболеванием.
Анализы также можно проводить после рождения для диагностики СМА у детей и взрослых.
Подробнее о тестах на SMA.
Как передается SMA
В большинстве случаев ребенок может родиться с СМА только в том случае, если у обоих родителей есть дефектный ген, вызывающий заболевание.
У родителей обычно не бывает SMA, которая известна как носитель. Примерно 1 из 40-60 человек является носителем основного дефектного гена, вызывающего СМА.
Если у 2 родителей-носителей есть ребенок, существует:
- 1 из 4 (25%) шансов, что их ребенок будет иметь SMA
- 1 из 2 (50%) шанс, что их ребенок будет носителем дефектного ген, но не будет иметь SMA
- 1 из 4 (25%) шанс, что их ребенок не будет иметь SMA и не будет носителем
Некоторые более редкие типы SMA наследуются немного по-другому или могут не передаваться на всех.
Поговорите с врачом, если у вас или вашего партнера есть семейная история СМА, и вы беспокоитесь, что ваши дети могут заболеть этим.
Подробнее о том, как СМА передается по наследству, можно узнать на веб-сайте Spinal Muscular Atrophy UK
Национальная служба регистрации врожденных аномалий и редких заболеваний
Если у вас или у вашего ребенка спинальная мышечная атрофия, ваша клиническая бригада передаст информацию о вас или вашем ребенке в Национальную службу регистрации врожденных аномалий и редких заболеваний (NCARDRS).
NCARDRS помогает ученым искать более эффективные способы предотвращения и лечения спинальной мышечной атрофии.Вы можете отказаться от регистрации в любое время.
Последняя проверка страницы: 4 мая 2020 г.
Срок следующего рассмотрения: 4 мая 2023 г.
: Механизмы и стратегии противодействия атрофии мышц | Журналы геронтологии: серия A
Аннотация
Размер скелетных мышц определяется рядом факторов, включая мышечную нагрузку, использование и регенеративную способность. Удивительно, но действия, которые могут способствовать росту мышц, не обязательно предотвращают потерю мышечной массы или атрофию.Это говорит о том, что дивергентные механизмы важны для поддержания мышечной массы в разных контекстах. При острой атрофии мышцы быстро теряют массу при отсутствии нагрузки, и эта реакция, по-видимому, включает активное удаление миоядер. Напротив, хроническая атрофия, такая как потеря мышечной массы, связанная со старением, связана с нарушениями восстановления мышц. В этом обзоре исследуются два контекста потери мышечной массы, чтобы определить, задействованы ли аналогичные процессы.
В этом обзоре исследуются два контекста потери мышечной массы, чтобы определить, задействованы ли аналогичные процессы.
КАКОВЫ потенциальные механизмы, лежащие в основе атрофии, регенерации и восстановления мышц? Ключ к этому вопросу может заключаться в сателлитных клетках (Sc), которые лежат на периферии мышечных волокон (1).Миофибриллы, будучи постмитотическими многоядерными клетками, зависят от синцития многих ядер. Миоядерный домен — это область волокна, которая поддерживается одним ядром; Экспрессия гена и синтез белка, которые происходят в домене, поддерживаются одним ядром (2). Если сохраняется экономичность волокна, этот миоядерный домен также необходимо поддерживать с течением времени. Когда СК активируются на периферии мышечного волокна во время роста или во время повреждения, они пролиферируют, дифференцируются, а затем сливаются с поврежденными или растущими миофибриллами.Добавление миоядер к существующим волокнам либо компенсирует потерю ядер в результате повреждения, либо поддерживает существующий миоядерный пул, обеспечивая повышенный синтез белка и, следовательно, более крупные мышечные волокна. Добавление новых ядер к мышечным волокнам поддерживает миоядерный домен. Атрофия мышц, в некотором смысле, является обратным процессом, поскольку при уменьшении размера волокна происходит систематическое устранение этих миоядер. Итак, в качестве первоначального вопроса мы спросили, являются ли мышечная атрофия и мышечная регенерация просто функциональными противоположностями друг друга, или они действительно не связаны между собой.Поскольку мы сосредоточились в первую очередь на стимулировании регенерации и восстановления мышц, мы начали проверять взаимосвязь между регенерацией и атрофией, исследуя факторы, которые способствуют регенерации мышц в контексте мышечной атрофии.
Существует два концептуально различных вида атрофии. Острая атрофия связана с неиспользованием, а хроническая атрофия связана со старением, которое может быть основной причиной саркопении. С возрастом могут возникать оба вида атрофии, и старение может нарушить процесс восстановления после состояния острой атрофии (3). Хотя между острой и хронической атрофией есть некоторое сходство, включая сопоставимое уменьшение мышечной массы и размера волокон, существуют также ключевые различия. Удельная сила (сила на площадь поперечного сечения) значительно уменьшается только при старении или хронической атрофии (4). Свойства волокон имеют тенденцию смещаться в сторону более быстрых типов волокон при острой атрофии, тогда как при хронической атрофии наблюдается уменьшение как общего количества волокон, так и избирательная атрофия самых быстрых и мощных типов волокон (5).
Хотя между острой и хронической атрофией есть некоторое сходство, включая сопоставимое уменьшение мышечной массы и размера волокон, существуют также ключевые различия. Удельная сила (сила на площадь поперечного сечения) значительно уменьшается только при старении или хронической атрофии (4). Свойства волокон имеют тенденцию смещаться в сторону более быстрых типов волокон при острой атрофии, тогда как при хронической атрофии наблюдается уменьшение как общего количества волокон, так и избирательная атрофия самых быстрых и мощных типов волокон (5).
Для наших исследований острой атрофии мы приняли модель подвешивания задних конечностей у мышей (6,7). В течение 2 недель подвешивания задних конечностей камбаловидная мышца, общая постуральная мышца, теряет за это время примерно 35-40% своей мышечной массы. Напротив, мышца длинного разгибателя пальцев (EDL), быстрая мышца, которая задействуется не так часто, как камбаловидная мышца, не демонстрирует такого же вида атрофии. На поперечных срезах, взятых из середины живота камбаловидной мышцы мыши в неподвешенном случае и после 14 дней подвешивания, наблюдается резкое уменьшение размера мышцы, пораженной этой неиспользованной атрофией (рис. 1).
1).
Основными датчиками изменений активности (и нагрузки) в мышцах являются комплекс интегрина и комплекс, связанный с дистрофином. Оба этих комплекса охватывают сарколемму, прикрепляясь к актиновому цитоскелету внутри клетки и к внеклеточному матриксу снаружи клетки. Силы, генерируемые внутри клетки, передаются через мембрану через эти комплексы. Оба эти комплекса не обладают собственными сигнальными способностями; однако кажется, что они опосредуют эти сигналы нагрузки через киназу фокальной адгезии (FAK) (8,9).FAK представляет собой нацеленную киназу, которая питается множеством различных путей, приводя к активации путей роста и предотвращению апоптоза. Активация рецептора инсулиноподобного фактора роста (IGF) -I также использует эти пути (10). В ненагруженной камбаловидной мышце общее количество FAK и его активность снижается (11). Другие белки, связанные с комплексом фокальной адгезии, также начинают подавлять регуляцию, включая винкулин и талин, которые подвергаются расщеплению (C. Morris, неопубликованные наблюдения).
Morris, неопубликованные наблюдения).
Хотя IGF-I способствует росту, он не может предотвратить потерю мышечной массы, связанную с неиспользованием.У трансгенных животных, которые экспрессируют высокие уровни IGF-I, наблюдается уменьшение мышечной массы после подвешивания задних конечностей, которое пропорционально наблюдаемому у нетрансгенных животных (12). Кроме того, системное введение гормона роста и IGF-I крысам не предотвращает атрофию неиспользования, связанную с этим подвешиванием задних конечностей (13). Однако добавление периодических упражнений к введению IGF-I в течение периода подвешивания задних конечностей может ослабить атрофический ответ.
Эти эксперименты предполагают, что активация рецептора IGF-I не приводит к предотвращению атрофии неиспользования.Отсутствие мышечной нагрузки каким-то образом блокирует активность рецептора IGF-I. Однако возвращение обеих мышечных нагрузок, возможно, через комплекс интегрина и комплекс дистрофина, позволяет IGF-I выполнять свою работу. Поэтому, чтобы предотвратить атрофию неиспользования, мы имитировали нагрузку на мышцы. Поскольку активация мембранных комплексов не блокирует потерю мышечной массы, мы заглянули внутрь цитозоля, чтобы найти, какие еще факторы мы могли бы использовать для блокирования мышечной атрофии. Хотя есть много факторов на выбор, включая участников сигнального каскада, большинство сигнальных белков участвует во многих путях.Следовательно, эффекты модификаций этих белков не будут ограничиваться блокированием мышечной атрофии, и они не могут обеспечить четкую проверку того, какой механизм лежит в основе процесса мышечной атрофии. Однако один белок — BCL-2 — выделяется тем, что является ключевым регулятором апоптоза.
Поэтому, чтобы предотвратить атрофию неиспользования, мы имитировали нагрузку на мышцы. Поскольку активация мембранных комплексов не блокирует потерю мышечной массы, мы заглянули внутрь цитозоля, чтобы найти, какие еще факторы мы могли бы использовать для блокирования мышечной атрофии. Хотя есть много факторов на выбор, включая участников сигнального каскада, большинство сигнальных белков участвует во многих путях.Следовательно, эффекты модификаций этих белков не будут ограничиваться блокированием мышечной атрофии, и они не могут обеспечить четкую проверку того, какой механизм лежит в основе процесса мышечной атрофии. Однако один белок — BCL-2 — выделяется тем, что является ключевым регулятором апоптоза.
При резком изменении уровня активности, особенно в модели атрофии неиспользования, усиление апоптоза связано с удалением миоядер из этих волокон (14,15). Поскольку было зарегистрировано, что апоптотические события происходят при атрофии неиспользования, блокирование этих событий может блокировать атрофию, связанную с неиспользованием. BCL-2 работает, блокируя высвобождение цитохрома С из митохондрий (16). Одним из аспектов провоцирования апоптоза является разобщение митохондрий; при расцеплении они высвобождают цитохром С, который запускает каскад событий, ведущих к апоптозу. Так что, если мы сможем предотвратить возникновение этого первоначального триггера апоптоза, имея в клетке высокие уровни агента, который может его блокировать, возможно, мы также сможем предотвратить атрофию неиспользования. Предварительные эксперименты подтверждают, что экспрессия BCL-2 может уменьшить потерю мышечной массы, связанную с неиспользованием.
BCL-2 работает, блокируя высвобождение цитохрома С из митохондрий (16). Одним из аспектов провоцирования апоптоза является разобщение митохондрий; при расцеплении они высвобождают цитохром С, который запускает каскад событий, ведущих к апоптозу. Так что, если мы сможем предотвратить возникновение этого первоначального триггера апоптоза, имея в клетке высокие уровни агента, который может его блокировать, возможно, мы также сможем предотвратить атрофию неиспользования. Предварительные эксперименты подтверждают, что экспрессия BCL-2 может уменьшить потерю мышечной массы, связанную с неиспользованием.
Старение мышц, модель хронической атрофии, приводит к потере около одной трети мышечной массы и силы у людей в возрасте от 30 до 80 лет (5). В продолжительности жизни мыши это представляет собой разницу между мышью в возрасте примерно 12 месяцев и мышью в возрасте примерно 2,5 лет. Удельная сила, то есть сила площади поперечного сечения, также уменьшается, и происходит потеря этих очень мощных типов волокон (4). Чтобы проверить, что вызывает снижение мышечной массы и функции, связанное со старением, мы применили те же тесты, что и при острой атрофии.
Чтобы проверить, что вызывает снижение мышечной массы и функции, связанное со старением, мы применили те же тесты, что и при острой атрофии.
IGF-I может увеличивать пролиферацию и регенерацию сателлитных клеток, а также может увеличивать синтез белка (17–20). Хотя сверхэкспрессия IGF-I не предотвращает острую атрофию, может ли его действие предотвратить хроническую атрофию? Поскольку потеря быстрых типов волокон связана с хронической атрофией, мы теперь нацелены на наши эксперименты на мышиной EDL, которая представляет собой хорошо охарактеризованную быструю мышцу идеального размера и формы для выполнения функционального анализа. Мы использовали энхансер промотора 1–3 легкой цепи миозина (который ограничивает экспрессию быстрыми скелетными мышцами) в аденоассоциированном вирусе для доставки гена IGF-I в EDL.Мы ввели мышей среднего возраста (т.е. мышей в возрасте 12–18 месяцев) и подождали, пока этим мышам не исполнилось 27 месяцев; Затем мы проанализировали их функциональные возможности, размер мышц и гистологию.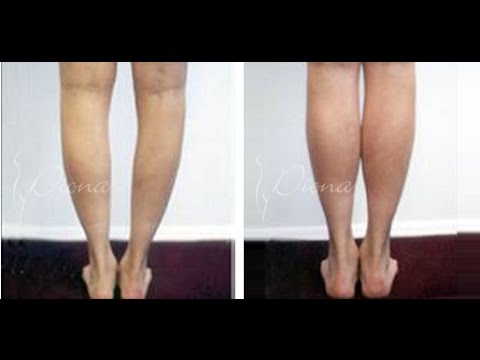 Мыши, которым вводили IGF-I, сохраняли как мышечную массу, так и удельную силу в течение периода старения по сравнению с контрольными конечностями без инъекции (21). Мы просто поддерживали мышечную функцию на уровне, который был в начале эксперимента.
Мыши, которым вводили IGF-I, сохраняли как мышечную массу, так и удельную силу в течение периода старения по сравнению с контрольными конечностями без инъекции (21). Мы просто поддерживали мышечную функцию на уровне, который был в начале эксперимента.
Как IGF-I работает в стареющих мышцах? По мере того как мышца с возрастом сокращается, сигналы восстановления, требующие регенерации мышц, уменьшаются.Повышенная экспрессия IGF-I усиливает путь регенерации, когда рецепторы IGF-I экспрессируются на активированных сателлитных клетках, тем самым увеличивая пролиферацию пула сателлитных клеток и увеличивая количество репарации, которая происходит во время процесса репарации и регенерации (Рисунок 2). Сверхэкспрессия IGF-I также предотвращает преимущественную потерю волокон типа IIb (быстрых), поскольку процент волокон IIb поддерживается на уровне около 65% в EDL мыши (21). Поскольку экспрессия IGF-I может предотвратить потерю мышечной функции, связанную со старением, ясно, что в основе хронической и острой атрофии лежат разные процессы. На данный момент неизвестно, способствует ли апоптоз и его профилактика хронической атрофии.
На данный момент неизвестно, способствует ли апоптоз и его профилактика хронической атрофии.
Чтобы показать, что IGF-I может усиливать процесс восстановления мышц, мы использовали трансгенную модель, экспрессирующую IGF-I, с использованием той же системы экспрессии, которая описана для вируса (22). Эту линию мышей скрестили с линией, в которой экспрессия репортерного гена регулировалась промотором, который временно включался только в активированных сателлитных клетках. Мышцы у старых мышей, несущих репортерный ген с экспрессией IGF-I и без нее, были повреждены, и во время выздоровления наблюдали за ними.У старых мышей без IGF-I наблюдалась нарушенная регенерация, тогда как у мышей с IGF-I восстановление улучшалось. Более того, только у мышей со сверхэкспрессией IGF-I были обнаружены активированные сателлитные клетки. Это подтверждает, что IGF-I усиливает процесс восстановления мышц. Это также предполагает, что, хотя IGF-I может не очень хорошо работать для предотвращения атрофии неиспользования, он может быть полезен в процессе восстановления, который происходит впоследствии.
Почему при старении происходит потеря волокон IIb? Одна из возможностей заключается в том, что это вопрос масштабирования, поскольку большие волокна имеют меньшее отношение площади поверхности к объему.Количество сократительного белка пропорционально объему мышцы, и этот сократительный белок создает силу, которая воздействует на относительно меньшую площадь поверхности, тем самым делая эти крупные волокна более восприимчивыми к повреждению. Преимущественное повреждение происходит в этих более крупных волокнах, которые оказываются волокнами IIb. Опять же, всегда будет происходить восстановление, но если повреждение будет значительным, эти мышечные волокна потеряют иннервацию. Реиннервация пула моторных нейронов будет происходить во время ремонта волокна.Однако существует также конкуренция между медленными мотонейронами и быстрыми мотонейронами, приводящими в действие эту мышцу. Исследование, проведенное Дезиприсом и Парри в 1990 году, показало, что если денервировать мышцу и позволить ей реиннервировать, медленные мотонейроны побеждают в этой гонке (23). Пул мотонейронов типа I (медленный) увеличивается после реинервации. В результате два различных процесса, возможно, приводят к уменьшению популяции волокон типа IIb в старых мышцах: во-первых, преимущественное повреждение крупных волокон и, во-вторых, преимущественная реиннервация медленными двигательными нейронами.Неясно, где IGF-I участвует в предотвращении быстрой потери клетчатки. Мы увеличили или позволили мышце восстанавливать себя еще быстрее, что, по нашему мнению, верно, или мы также увеличиваем способность быстрых мотонейронов к более полной иннервации?
Пул мотонейронов типа I (медленный) увеличивается после реинервации. В результате два различных процесса, возможно, приводят к уменьшению популяции волокон типа IIb в старых мышцах: во-первых, преимущественное повреждение крупных волокон и, во-вторых, преимущественная реиннервация медленными двигательными нейронами.Неясно, где IGF-I участвует в предотвращении быстрой потери клетчатки. Мы увеличили или позволили мышце восстанавливать себя еще быстрее, что, по нашему мнению, верно, или мы также увеличиваем способность быстрых мотонейронов к более полной иннервации?
Почему при острой атрофии происходит потеря медленных волокон? Сократительные белки и профили экспрессии генов, которые были выполнены совсем недавно Cros и коллегами (24), показывают, что эти профили экспрессии смещаются в сторону быстрых свойств волокон.Сдвиги волокон при острой атрофии, вероятно, зависят от активности. Мы знаем из нескольких лет работы Петте и Врбовой, что тоническая стимуляция способствует медленной экспрессии генов волоконного типа (25).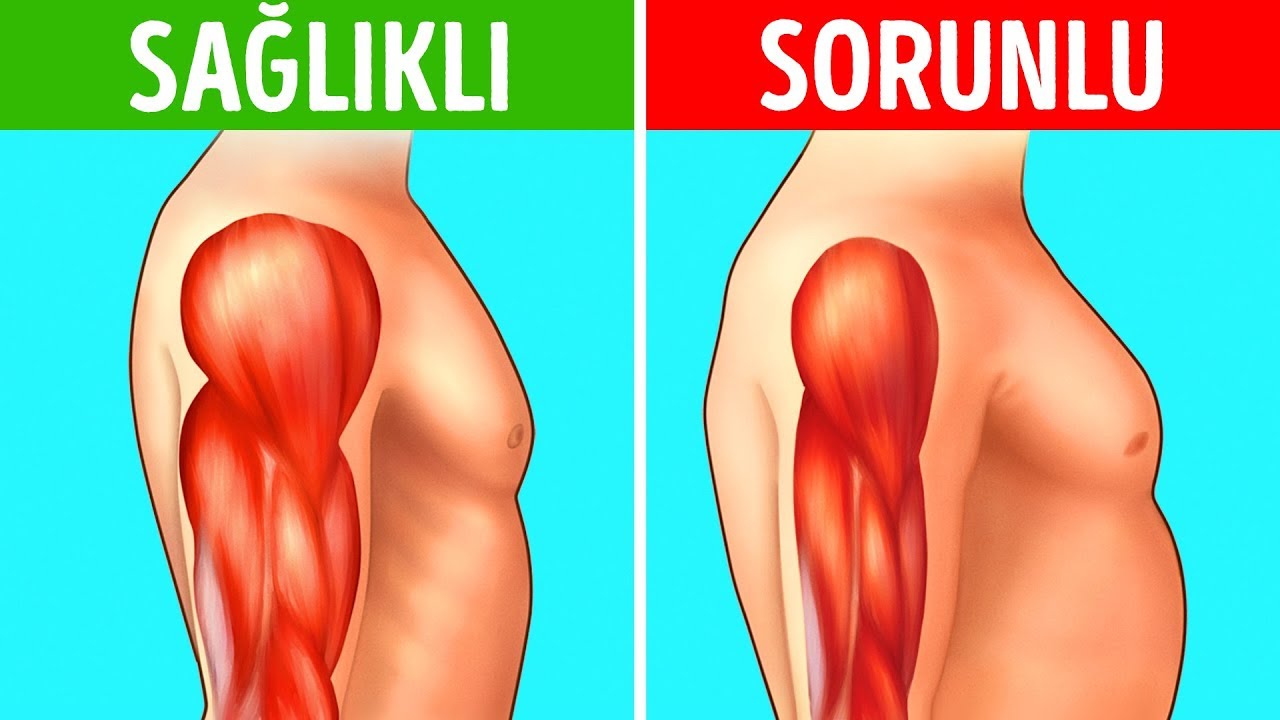 В камбаловидной мышце мыши наличие тонической активности, вероятно, поддерживает профиль медленного типа волокон. Однако при атрофии неиспользования наблюдается меньшая нервная активность. Следовательно, при недостатке нервной деятельности или отсутствии тонической стимуляции мышцы могут сдвинуться к своему исходному состоянию, то есть перейти в сторону быстрого типа волокон.Итак, опять же, это два очень разных процесса, которые происходят при острой и хронической атрофии: нарушение восстановления происходит с процессом старения, а отсутствие тонической активности способствует сдвигу волокон при острой атрофии.
В камбаловидной мышце мыши наличие тонической активности, вероятно, поддерживает профиль медленного типа волокон. Однако при атрофии неиспользования наблюдается меньшая нервная активность. Следовательно, при недостатке нервной деятельности или отсутствии тонической стимуляции мышцы могут сдвинуться к своему исходному состоянию, то есть перейти в сторону быстрого типа волокон.Итак, опять же, это два очень разных процесса, которые происходят при острой и хронической атрофии: нарушение восстановления происходит с процессом старения, а отсутствие тонической активности способствует сдвигу волокон при острой атрофии.
Возможные методы лечения острой атрофии могут включать антиапоптотические препараты, которые могут помочь предотвратить удаление миоядер (26). Другая возможность, которая не была изучена, — это имитация мышечной нагрузки. Если мы каким-то образом заставим мышцу нагружаться, возможно, потеря мышечной массы будет предотвращена.IGF-I совсем не помогает при острой атрофии, но может помочь в процессе выздоровления.
Атрофия мышц — это активный или пассивный процесс? Путь прямого роста фактически прекращается, поскольку через пути IGF-I происходит меньшая активация, и было показано, что IGF-I неэффективен в предотвращении этой атрофии неиспользования. Это говорит о том, что атрофия неиспользования — это пассивный процесс. Однако, по-видимому, также существует активный процесс, который направляет систематическое удаление миоядер путем апоптоза.Следовательно, может потребоваться комбинация методов лечения для предотвращения потери мышечной массы, связанной с бездействием и старением.
Рисунок 1.
Ответ скелетных мышц мыши на подвешивание задних конечностей. A , Относительная мышечная масса длинного разгибателя пальцев (EDL, сплошная линия) и камбаловидной мышцы (SOL, пунктирная линия) как функция продолжительности подвешивания задних конечностей. Масса мышц, которые обычно активируются часто, такие как камбаловидная мышца, более восприимчивы к разгрузке, чем те, которые задействуются нечасто, такие как EDL. B , Поперечные срезы камбаловидной мышцы без подвешивания задних конечностей (слева) или после 14 дней подвешивания (справа)
B , Поперечные срезы камбаловидной мышцы без подвешивания задних конечностей (слева) или после 14 дней подвешивания (справа)
Рисунок 1.
Реакция скелетных мышц мыши на подвешивание задних конечностей. A , Относительная мышечная масса длинного разгибателя пальцев (EDL, сплошная линия) и камбаловидной мышцы (SOL, пунктирная линия) как функция продолжительности подвешивания задних конечностей. Масса мышц, которые обычно активируются часто, такие как камбаловидная мышца, более восприимчивы к разгрузке, чем те, которые задействуются нечасто, такие как EDL. B , Поперечные срезы камбаловидной мышцы без подвешивания задних конечностей (слева) или после 14 дней подвешивания (справа)
Рисунок 2.
Схема регенерации и восстановления скелетных мышц. Клетки-сателлиты на периферии мышечных волокон активируются связыванием фактора роста гепатоцитов (HGF) с его рецептором (c-met), после чего клетки-сателлиты экспрессируют рецепторы инсулиноподобного фактора роста (IGF) -I (IGFR). Высокие уровни IGF-I могут улучшить восстановление мышц, стимулируя пролиферацию и дифференцировку сателлитных клеток
Высокие уровни IGF-I могут улучшить восстановление мышц, стимулируя пролиферацию и дифференцировку сателлитных клеток
Рисунок 2.
Схема регенерации и восстановления скелетных мышц. Клетки-сателлиты на периферии мышечных волокон активируются связыванием фактора роста гепатоцитов (HGF) с его рецептором (c-met), после чего клетки-сателлиты экспрессируют рецепторы инсулиноподобного фактора роста (IGF) -I (IGFR). Высокий уровень IGF-I может улучшить восстановление мышц, стимулируя пролиферацию и дифференциацию сателлитных клеток.
Адрес для корреспонденции Элизабет Бартон, PhD, кафедра анатомии и клеточной биологии, Школа стоматологической медицины Пенсильванского университета, 4001 Spruce Street, Philadelphia, PA 19104 .Эл. Почта: [email protected]
Список литературы
1Мауро А. Сателлитная клетка волокон скелетных мышц.
Дж Биофизика.
.1961
;9
:493
-498,2 Аллен Д. Л., Рой Р.Р., Эджертон В.Р. Миоядерные домены в адаптации и заболеваниях мышц.
Л., Рой Р.Р., Эджертон В.Р. Миоядерные домены в адаптации и заболеваниях мышц.
Мышечный нерв.
.1999
;22
:1350
-1360,3Заржевский Н., Кармели Э., Фукс Д., Коулман Р., Стейн Х., Резник А.З.Восстановление мышц старых крыс после иммобилизации задних конечностей внешней фиксацией ухудшается по сравнению с таковыми у молодых крыс.
Exper Gerontol.
.2001
;36
:125
-140,4Brooks SV, Faulkner JA. Сократительные свойства скелетных мышц молодых, взрослых и старых мышей.
J Physiol.
.1988
;404
:71
-82,5Lexell J. Старение и мышцы человека: наблюдения из Швеции.
Can J Appl Physiol.
.1993
;18
:2
-18,6Musacchia XJ, Steffen JM, Fell RD. Атрофия скелетных мышц из-за неиспользования: модели на животных.
Exerc Sport Sci Rev.
.1988
;16
:61
-87,7 Thomason DB, стенд FW. Атрофия камбаловидной мышцы из-за разгрузки задней конечности.
J Appl Physiol.
.1990
;68
:1
-12,8David FS, Zage PE, Marcantonio EE. Интегрины взаимодействуют с очаговыми спайками множеством различных путей.
J Cell Physiol.
.1999
;181
:74
-82,9Schlaepfer DD, Hauck CR, Sieg DJ. Передача сигналов через киназу фокальной адгезии.
Progr Biophys Molec Biol.
.1997
;71
:435
-478,10Барон В., Каллея В., Феррари П., Аленгрин Ф. Ван Обберген Е. Киназа фокальной адгезии p125Fak является субстратом для рецепторов тирозинкиназы инсулина и инсулиноподобного фактора роста I.
J. Biol Chem.
.1998
;273
:7162
-7168.11Gordon SE, Fluck M, стенд FW. Пластичность скелетных, сердечных и гладких мышц: выбранный вклад: киназа фокальной адгезии скелетных мышц, паксиллин и фактор ответа сыворотки зависят от нагрузки.
J Appl Physiol.
.2001
;90
:1174
-1183.12 Criswell DS, Carson JA, Booth FW. Регулирование экспрессии гена сократительного белка в ненагруженных скелетных мышцах мышей.
Регулирование экспрессии гена сократительного белка в ненагруженных скелетных мышцах мышей.
J Gravit Physiol.
.1996
;3
:58
-60,13Аллен Д.Л., Линдерман Дж.К., Рой Р.Р., Гринделанд Р.Э., Мукку В.Р., Эдгертон В.Р. Гормон роста / IGF-I и / или упражнения с сопротивлением поддерживают количество миоядерных ядер в невзвешенных мышцах задних конечностей.
J Appl Physiol.
.1997
;83
:1857
-1861,14Аллен Д.Л., Линдерман Дж. К., Рой Р. Р. и др. Апоптоз: механизм, способствующий ремоделированию скелетных мышц в ответ на разгрузку задних конечностей.
Am J Physiol.
.1997
;273
:C579
-C587.15Аллен Д.Л., Монк С.Р., Талмадж Р.Дж., Рой Р.Р., Эдгертон В.Р. Пластичность миоядерного числа в гипертрофированных и атрофированных волокнах скелетных мышц млекопитающих.
J Appl Physiol.
.1995
;78
:1969
-1976,16 Адамс Дж. М., Кори С. Семейство белков Bcl-2: арбитры выживания клеток.
Наука.
.1998
;281
:1322
-1326.17Адамс Г.Р., МакКью С.А. Локальное введение IGF-I приводит к гипертрофии скелетных мышц у крыс.
J Appl Physiol.
.1998
;84
:1716
-1722.18Бартон-Дэвис Е.Р., Шотурма Д.И., Суини Х.Л. Вклад сателлитных клеток в гипертрофию скелетных мышц, индуцированную IGF-I.
Acta Physiol Scand.
.1999
;167
:301
-305,19Чакраварти М.В., Абраха Т.В., Шварц Р.Дж., Фиоротто М.Л., стенд FW.Инсулиноподобный фактор роста-I увеличивает продолжительность репликативной продолжительности жизни in vitro сателлитных клеток скелетных мышц за счет усиления прогрессирования клеточного цикла G 1 / S посредством активации пути передачи сигналов фосфатидилинозитол-3′-киназы / Akt.
J. Biol Chem.
.2000
;275
:35942
-35952.20 Чакраварти М.В., Дэвис Б.С., Бут Ф.В. IGF-I восстанавливает потенциал пролиферации сателлитных клеток в иммобилизованных старых скелетных мышцах.
J Appl Physiol.
.2000
;89
:1365
-1379.21Бартон-Дэвис Э. Р., Шотурма Д. И., Мусаро А., Розенталь Н., Суини Х. Л.. Опосредованная вирусами экспрессия инсулиноподобного фактора роста I блокирует связанную со старением потерю функции скелетных мышц.
Proc Natl Acad Sci U S A.
.1998
;95
:15603
-15607,22Musaro A, McCullagh K, Paul A и др. Локальная экспрессия трансгена IGF-I поддерживает гипертрофию и регенерацию стареющих скелетных мышц.
Nat Genet.
.2001
;27
:195
-200,23Desypris G, Parry DJ. Относительная эффективность медленных и быстрых альфа-мотонейронов для реиннервации камбаловидной мышцы мыши.
Am J Physiol Cell Physiol.
.1990
;258
:C62
-C70.24Cros N, Ткаченко А.В., Пизани Д.Ф. и др. Анализ измененной экспрессии гена в камбаловидной мышце крысы, атрофированной из-за неиспользования.
J Cell Biochem.
.
2001
;83
:508
-519.25Петте Д., Врбова Г. Что говорит нам хроническая электростимуляция о пластичности мышц?
Мышечный нерв.
.1999
;22
:666
-677.26Leeuwenburgh C. Роль апоптоза в саркопении. J Gerontol Med Sci. В печати.
Американское геронтологическое общество
Во время атрофии мышц толстые, но не тонкие компоненты филаментов разрушаются за счет MuRF1-зависимого убиквитилирования | Журнал клеточной биологии
Потеря миофибриллярных белков является признаком атрофии мышц.Экспрессия мышечной RING-finger 1 (MuRF1), убиквитинлигазы, заметно индуцируется во время атрофии, а делеция MuRF1 снижает мышечное истощение. Мы создали мышей, экспрессирующих мутантный MuRF1 с делецией кольца, который связывается, но не может убиквитилировать субстраты. Масс-спектрометрия связанных белков в денервированных мышцах идентифицировала многие миофибриллярные компоненты. После денервации или голодания атрофированные мышцы демонстрируют потерю миозин-связывающего протеина C (MyBP-C) и легких цепей миозина 1 и 2 (MyLC1 и MyLC2) из миофибрилл до любого измеримого уменьшения тяжелой цепи миозина (MyHC).Их избирательная потеря требует MuRF1. MyHC защищен от убиквитилирования в миофибриллах ассоциированными белками, но в конечном итоге подвергается MuRF1-зависимой деградации. Напротив, MuRF1 убиквитилирует MyBP-C, MyLC1 и MyLC2 даже в миофибриллах. Поскольку эти белки стабилизируют толстые филаменты, их избирательное убиквитилирование может облегчить разборку толстых филаментов. Однако компоненты тонкой нити уменьшаются по механизму, не требующему MuRF1.
После денервации или голодания атрофированные мышцы демонстрируют потерю миозин-связывающего протеина C (MyBP-C) и легких цепей миозина 1 и 2 (MyLC1 и MyLC2) из миофибрилл до любого измеримого уменьшения тяжелой цепи миозина (MyHC).Их избирательная потеря требует MuRF1. MyHC защищен от убиквитилирования в миофибриллах ассоциированными белками, но в конечном итоге подвергается MuRF1-зависимой деградации. Напротив, MuRF1 убиквитилирует MyBP-C, MyLC1 и MyLC2 даже в миофибриллах. Поскольку эти белки стабилизируют толстые филаменты, их избирательное убиквитилирование может облегчить разборку толстых филаментов. Однако компоненты тонкой нити уменьшаются по механизму, не требующему MuRF1.
Атрофия скелетных мышц возникает при неиспользовании или денервации и системно при голодании и при многих болезненных состояниях, включая рак кахексию, сепсис, почечную и сердечную недостаточность (Lecker et al., 1999, 2006; Джекман и Кандарян, 2004 г.![]() ). Такая быстрая потеря мышечной массы в основном происходит из-за ускоренной деградации белка, хотя в системных катаболических состояниях синтез белка также снижается (Jackman and Kandarian, 2004; Lecker et al., 2006; Sacheck et al., 2007). Обычно белки, составляющие миофибриллы, являются одними из самых стабильных в организме. Однако во время атрофии миофибриллярный аппарат, который включает не менее 60% мышечных белков, уменьшается в массе в большей степени, чем растворимые компоненты (Munoz et al., 1993) и приводит к снижению прочности. Настоящие исследования были предприняты для понимания механизмов, ответственных за эту ускоренную деградацию миофибриллярных белков.
). Такая быстрая потеря мышечной массы в основном происходит из-за ускоренной деградации белка, хотя в системных катаболических состояниях синтез белка также снижается (Jackman and Kandarian, 2004; Lecker et al., 2006; Sacheck et al., 2007). Обычно белки, составляющие миофибриллы, являются одними из самых стабильных в организме. Однако во время атрофии миофибриллярный аппарат, который включает не менее 60% мышечных белков, уменьшается в массе в большей степени, чем растворимые компоненты (Munoz et al., 1993) и приводит к снижению прочности. Настоящие исследования были предприняты для понимания механизмов, ответственных за эту ускоренную деградацию миофибриллярных белков.
Недавние открытия показывают, что эти различные типы атрофии активируют общую серию биохимических и транскрипционных адаптаций (Sacheck et al., 2007), что приводит к усилению деградации белков как протеасомной, так и лизосомной системами (Mammucari et al. , 2007; Чжао и др., 2007). Распад миофибриллярных компонентов во время атрофии опосредуется убиквитин-протеасомным путем (Solomon and Goldberg, 1996). Несмотря на эту быструю деградацию различных мышечных белков во время атрофии, заметно индуцируются две мышечно-специфичные убиквитинлигазы (E3s), мышечный RING-finger 1 (MuRF1) и атрогин-1 / MAFbx; а у мышей с нокаутом, лишенных какого-либо фермента, быстрая потеря мышечной массы при денервации значительно снижается (Bodine et al., 2001; Gomes et al., 2001). На сегодняшний день неясно, убиквитилируют ли эти E3 многие компоненты мышц или только ключевые регуляторные, разрушение которых способствует распаду других белков. MuRF1 представляет собой мономерный E3, принадлежащий к семейству трехчастных мотивов лигаз, которые содержат трехкомпонентный домен RING: B-box: coiled-coil, который важен для конъюгации убиквитина. Два близких гомолога MuRF1 (MuRF2 и MuRF3) присутствуют в нормальной мышце и, по-видимому, важны для поддержания нормальной сократительной функции (Fielitz et al.
, 2007; Чжао и др., 2007). Распад миофибриллярных компонентов во время атрофии опосредуется убиквитин-протеасомным путем (Solomon and Goldberg, 1996). Несмотря на эту быструю деградацию различных мышечных белков во время атрофии, заметно индуцируются две мышечно-специфичные убиквитинлигазы (E3s), мышечный RING-finger 1 (MuRF1) и атрогин-1 / MAFbx; а у мышей с нокаутом, лишенных какого-либо фермента, быстрая потеря мышечной массы при денервации значительно снижается (Bodine et al., 2001; Gomes et al., 2001). На сегодняшний день неясно, убиквитилируют ли эти E3 многие компоненты мышц или только ключевые регуляторные, разрушение которых способствует распаду других белков. MuRF1 представляет собой мономерный E3, принадлежащий к семейству трехчастных мотивов лигаз, которые содержат трехкомпонентный домен RING: B-box: coiled-coil, который важен для конъюгации убиквитина. Два близких гомолога MuRF1 (MuRF2 и MuRF3) присутствуют в нормальной мышце и, по-видимому, важны для поддержания нормальной сократительной функции (Fielitz et al. , 2007; Witt et al., 2008). MuRF1, с другой стороны, необходим для быстрой атрофии, но не для нормального роста мышц.
, 2007; Witt et al., 2008). MuRF1, с другой стороны, необходим для быстрой атрофии, но не для нормального роста мышц.
В настоящих исследованиях мы использовали мышей, у которых отсутствует RING-домен MuRF1, чтобы выяснить роль его лигазной активности в атрофии денервации. Хотя сообщалось о двух субстратах для MuRF1, сердечного тропонина-I (Kedar et al., 2004) и тяжелой цепи миозина (MyHC) (Clarke et al., 2007), роль MuRF1 в разрушении специфических миофибриллярных или растворимых белков в скелетных тканях мышца неизвестна.В частности, неясно, может ли MuRF1 или любой E3 действовать на белки внутри миофибриллы, которая представляет собой высокоорганизованную жесткую структуру, или существуют ли механизмы для высвобождения белков из саркомеров перед убиквитин-зависимой деградацией. Модельные исследования мышечных экстрактов продемонстрировали быстрый гидролиз очищенного актина и миозина посредством убиквитин-протеасомного пути (Solomon and Goldberg, 1996). Однако эти белки были намного более стабильными, когда они были связаны друг с другом в актомиозиновом комплексе или интактных миофибриллах.Эти находки открывают возможность того, что система убиквитин-протеасома не может сама по себе разрушать компоненты интактных миофибрилл, а составляющие белки должны высвобождаться с помощью некоторого механизма, чтобы быть субстратами для деградации (Solomon and Goldberg, 1996). Следовательно, несколько групп представили доказательства того, что кальпаин (Tidball and Spencer, 2002) или каспаза (Du et al., 2004) могут первоначально расщеплять миофибриллярные компоненты, тем самым ускоряя разборку и деградацию системой убиквитин-протеасома.Однако прямых доказательств функционирования этих моделей в атрофированных мышцах нет. Альтернативно, E3 может действовать непосредственно на определенные компоненты миофибриллы, способствуя ее разборке и деградации во время атрофии. Здесь мы представляем доказательства такой роли MuRF1.
Однако эти белки были намного более стабильными, когда они были связаны друг с другом в актомиозиновом комплексе или интактных миофибриллах.Эти находки открывают возможность того, что система убиквитин-протеасома не может сама по себе разрушать компоненты интактных миофибрилл, а составляющие белки должны высвобождаться с помощью некоторого механизма, чтобы быть субстратами для деградации (Solomon and Goldberg, 1996). Следовательно, несколько групп представили доказательства того, что кальпаин (Tidball and Spencer, 2002) или каспаза (Du et al., 2004) могут первоначально расщеплять миофибриллярные компоненты, тем самым ускоряя разборку и деградацию системой убиквитин-протеасома.Однако прямых доказательств функционирования этих моделей в атрофированных мышцах нет. Альтернативно, E3 может действовать непосредственно на определенные компоненты миофибриллы, способствуя ее разборке и деградации во время атрофии. Здесь мы представляем доказательства такой роли MuRF1.
Миофибриллы состоят из точно выровненных систем филаментов, расположенных в повторяющихся единицах саркомеров (Clark et al.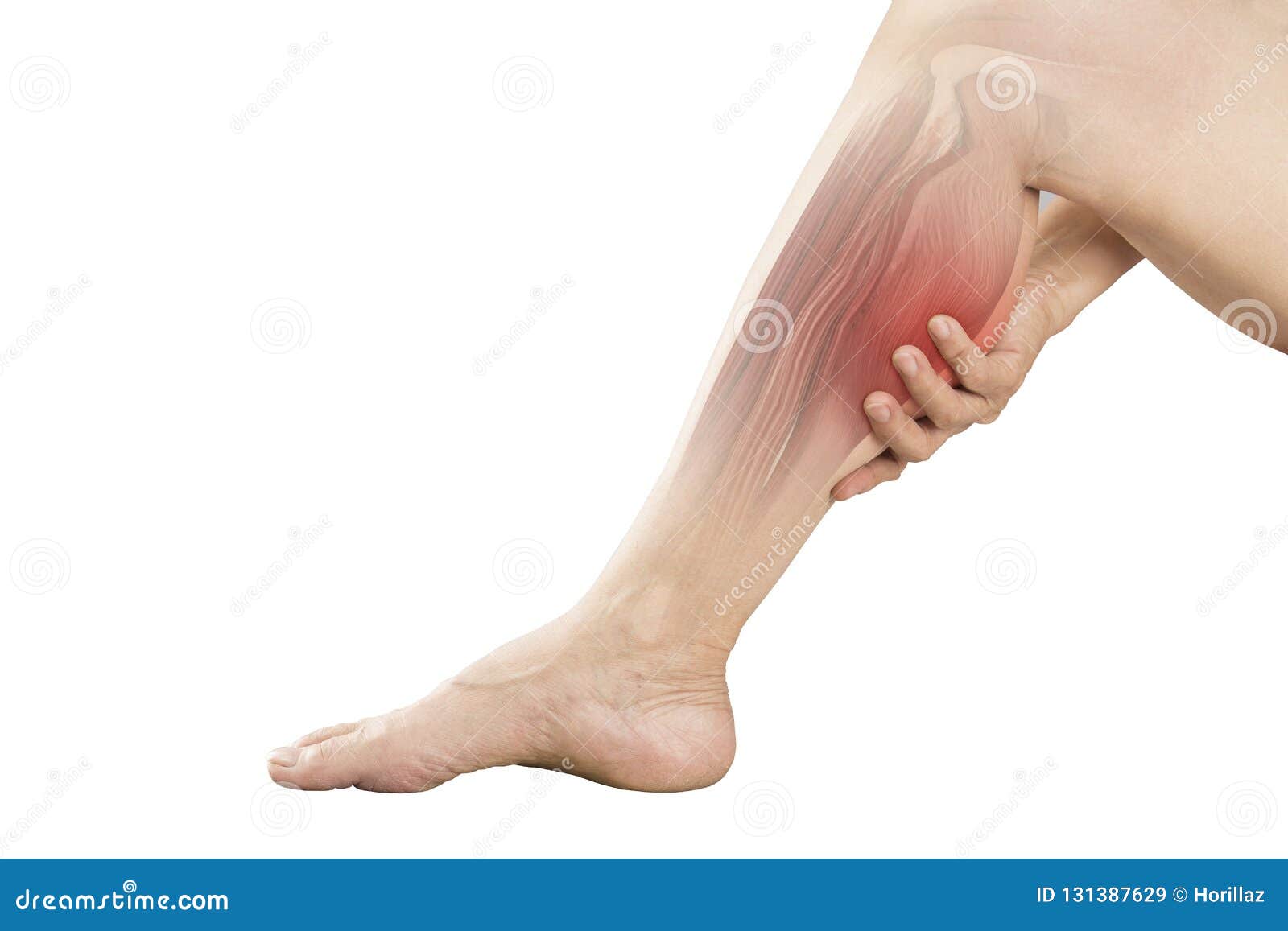 , 2002), основными составляющими которых являются миозин в толстом филаменте и актин в тонком филаменте.Молекула миозина составляет 50% миофибриллярной массы и содержит две тяжелые цепи, две регуляторные легкие цепи (MyLC2) и две основные легкие цепи (MyLC1). Эти легкие цепи связываются с глобулярными головками миозина, которые включают поперечные мостики, которые отходят от толстых нитей, чтобы взаимодействовать с актином и генерировать силу за счет гидролиза АТФ. Несколько наблюдений in vivo и с изолированными миофибриллами показывают, что MyLC1 и MyLC2 необходимы для стабилизации толстых филаментов и нормальной сократимости.Потеря MyLC1 или MyLC2 у людей (Rottbauer et al., 2006) или их гомологов у рыбок данио (Chen et al., 2008) приводит к нарушению сократимости и поразительной дезорганизации толстых нитей.
, 2002), основными составляющими которых являются миозин в толстом филаменте и актин в тонком филаменте.Молекула миозина составляет 50% миофибриллярной массы и содержит две тяжелые цепи, две регуляторные легкие цепи (MyLC2) и две основные легкие цепи (MyLC1). Эти легкие цепи связываются с глобулярными головками миозина, которые включают поперечные мостики, которые отходят от толстых нитей, чтобы взаимодействовать с актином и генерировать силу за счет гидролиза АТФ. Несколько наблюдений in vivo и с изолированными миофибриллами показывают, что MyLC1 и MyLC2 необходимы для стабилизации толстых филаментов и нормальной сократимости.Потеря MyLC1 или MyLC2 у людей (Rottbauer et al., 2006) или их гомологов у рыбок данио (Chen et al., 2008) приводит к нарушению сократимости и поразительной дезорганизации толстых нитей.
Кроме того, саркомер содержит множество структурных и регуляторных белков, которые способствуют целостности и стабильности миофибрилл.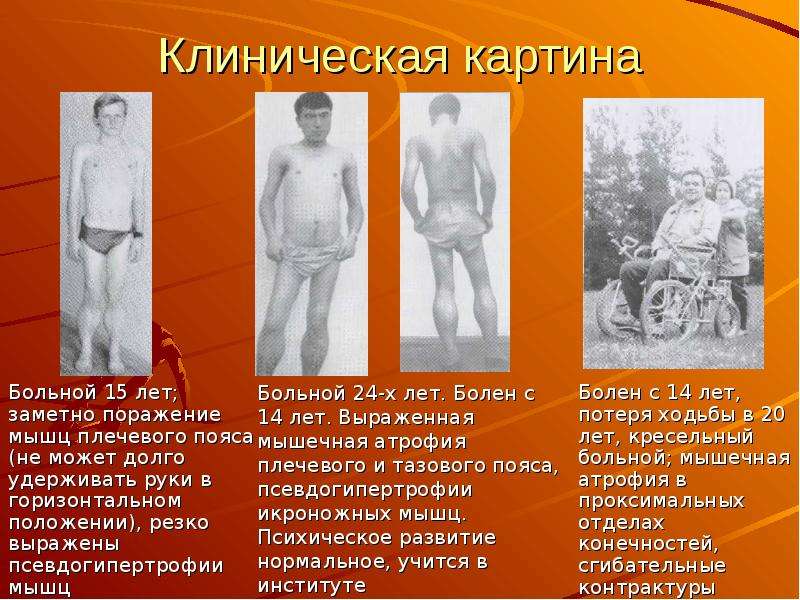 Например, миозин-связывающий протеин C (MyBP-C) является компонентом толстого филамента и распределяется через каждые 43 нм в C-зоне саркомера, где находятся поперечные мостики (Offer et al., 1972). Трансгенные мыши, экспрессирующие усеченный MyBP-C, у которого отсутствует сайт связывания миозина в сердечной мышце, обнаруживают значительную дезорганизацию саркомера (Yang et al., 1998). Поскольку MyBP-C, MyLC1 и MyLC2 необходимы для стабильности саркомеров, их избирательная потеря и быстрое убиквитилирование, которое мы демонстрируем здесь, вероятно, будут механизмом, способствующим разборке миофибрилл во время атрофии.
Например, миозин-связывающий протеин C (MyBP-C) является компонентом толстого филамента и распределяется через каждые 43 нм в C-зоне саркомера, где находятся поперечные мостики (Offer et al., 1972). Трансгенные мыши, экспрессирующие усеченный MyBP-C, у которого отсутствует сайт связывания миозина в сердечной мышце, обнаруживают значительную дезорганизацию саркомера (Yang et al., 1998). Поскольку MyBP-C, MyLC1 и MyLC2 необходимы для стабильности саркомеров, их избирательная потеря и быстрое убиквитилирование, которое мы демонстрируем здесь, вероятно, будут механизмом, способствующим разборке миофибрилл во время атрофии.
В этих исследованиях мы использовали биохимические и генетические подходы для выяснения in vivo вклада лигазной активности MuRF1 в атрофический процесс и для выяснения роли MuRF1 в разборке миофибриллярного аппарата у взрослых мышей.В частности, мы исследовали (1) может ли какой-либо миофибриллярный белок избирательно теряться во время атрофии денервации, (2) важно ли убиквитилирование MuRF1 для деградации различных миофибриллярных белков, теряемых во время атрофии, (3) может ли MuRF1 убиквитилировать регуляторные белки, потеря которых может способствовать разрушению других миофибриллярных компонентов, и (4) могут ли миофибриллярные белки, которые разрушаются по MuRF1-зависимому механизму, убиквитилировать внутри миофибриллы, в экстрагированном актомиозиновом комплексе или только при выделении и очистке. Эти исследования неожиданно демонстрируют, что существуют разные механизмы деградации компонентов толстых и тонких филаментов и что определенные ассоциированные с толстыми филаментами белки теряются по-разному во время атрофии и выборочно убиквитилируются внутри интактной миофибриллы. Их дифференциальная потеря может представлять собой критический шаг, который способствует дальнейшему убиквитилированию и деградации других миофибриллярных компонентов.
Эти исследования неожиданно демонстрируют, что существуют разные механизмы деградации компонентов толстых и тонких филаментов и что определенные ассоциированные с толстыми филаментами белки теряются по-разному во время атрофии и выборочно убиквитилируются внутри интактной миофибриллы. Их дифференциальная потеря может представлять собой критический шаг, который способствует дальнейшему убиквитилированию и деградации других миофибриллярных компонентов.
Чтобы понять роль MuRF1 в мышечной атрофии, мы стремились идентифицировать его субстраты.Растворимые экстракты икроножных мышц нормальных мышей (3000 супернатант г ) инкубировали с иммобилизованным GST-MuRF1 для выделения тех мышечных белков, которые имеют высокое сродство к MuRF1. Полученные осадки тщательно промывали буфером, содержащим 500 мМ NaCl, для удаления неспецифических или слабо связанных белков. Белки на гранулах анализировали с помощью SDS-PAGE (фиг. S1) и масс-спектрометрии. Были идентифицированы многие миофибриллярные компоненты, особенно компоненты толстых нитей и области M-линии (Таблица I), включая миозин-связывающий белок C (MyBP-C, также называемый C-белком), эссенциальную легкую цепь миозина (MyLC1) и регуляторные легкая цепь (MyLC2), изоформа тяжелой цепи миозина (MyHC) типа II, миомезин 1 и филамин C (также называемый филамин 2, γ-филамин или ABP-L), компонент, локализованный на Z-диске.Это связывание MyLC2 согласуется с результатами скрининга библиотеки мышечной кДНК на MuRF1-связывающие белки с помощью дрожжевого двугибридного анализа (Witt et al., 2005). Более того, было показано, что MyHC (Clarke et al., 2007) убиквитилирован очищенным MuRF1. Мы не смогли осаждать тропонин-I из экстрактов скелетных мышц с помощью MuRF1, хотя, как сообщается, сердечный тропонин-I является субстратом MuRF1 in vivo (Kedar et al., 2004) и легко убиквитилируется очищенным MuRF1 (Kim et al. , 2007).
S1) и масс-спектрометрии. Были идентифицированы многие миофибриллярные компоненты, особенно компоненты толстых нитей и области M-линии (Таблица I), включая миозин-связывающий белок C (MyBP-C, также называемый C-белком), эссенциальную легкую цепь миозина (MyLC1) и регуляторные легкая цепь (MyLC2), изоформа тяжелой цепи миозина (MyHC) типа II, миомезин 1 и филамин C (также называемый филамин 2, γ-филамин или ABP-L), компонент, локализованный на Z-диске.Это связывание MyLC2 согласуется с результатами скрининга библиотеки мышечной кДНК на MuRF1-связывающие белки с помощью дрожжевого двугибридного анализа (Witt et al., 2005). Более того, было показано, что MyHC (Clarke et al., 2007) убиквитилирован очищенным MuRF1. Мы не смогли осаждать тропонин-I из экстрактов скелетных мышц с помощью MuRF1, хотя, как сообщается, сердечный тропонин-I является субстратом MuRF1 in vivo (Kedar et al., 2004) и легко убиквитилируется очищенным MuRF1 (Kim et al. , 2007).
Чтобы подтвердить, что эти связывающие MuRF1 белки (таблица I) действительно являются потенциальными субстратами, мы повторили выделение белков вместе с очищенным GST-MuRF1 из нормальных икроножных мышц и впоследствии проверили, могут ли эти связанные компоненты быть убиквитилированы после добавления E1, Ubch2. , и His-убиквитин.Полиубиквитилированные белки, меченные His, очищали с помощью никелевой колонки, и масс-спектрометрический анализ идентифицировал все эти миофибриллярные белки как виды, убиквитилированные MuRF1 (таблица I).
, и His-убиквитин.Полиубиквитилированные белки, меченные His, очищали с помощью никелевой колонки, и масс-спектрометрический анализ идентифицировал все эти миофибриллярные белки как виды, убиквитилированные MuRF1 (таблица I).
Для анализа регуляции экспрессии Myc-MuRF1 и Myc-ΔR-MuRF1 во время атрофии мы односторонне разрезали седалищный нерв в двух группах мышей. Через 10 дней животных умерщвляли и получали экстракты икроножных мышц.Заметную индукцию белка Myc-MuRF1 в этих лизатах наблюдали с помощью вестерн-блоттинга и напоминали индукцию MuRF1 у мышей WT (фиг. 1B). Интересно, что содержание Myc-ΔR-MuRF1 в денервированных мышцах было выше, чем содержание MuRF1 у мышей WT (рис. 1 D), вероятно, потому что, как и многие E3, MuRF1 аутубиквитилирует и в отсутствие домена безымянного пальца белок неактивен и, следовательно, намного более стабилен (Qiu and Goldberg, 2002). Несмотря на то, что в нем отсутствует домен RING (55 аминокислот), экспрессированный белок Myc-ΔR-MuRF1 был очень похож по размеру на эндогенный MuRF1, поскольку он содержит тег Myc (36 аминокислот). Как и ожидалось, размер белка Myc-MuRF1 был больше, чем размер Myc-ΔR-MuRF1 из-за присутствия домена RING.
Как и ожидалось, размер белка Myc-MuRF1 был больше, чем размер Myc-ΔR-MuRF1 из-за присутствия домена RING.
Чтобы определить, была ли активность убиквитинлигазы MuRF1 существенной для атрофии и деградации определенных миофибриллярных белков, мы проанализировали скорость атрофии денервации, чтобы узнать, снижает ли удаление безымянного пальца MuRF1 атрофию таким же образом, как и полная делеция MuRF1 (Bodine et al. ., 2001). Односторонняя атрофия мышц задних конечностей была вызвана разрезанием правого седалищного нерва в MuRF1 — / — , MuRF1 myc / myc и ΔR-MuRF1 myc / myc в трех отдельных экспериментах с Однопометники дикого типа из каждой когорты в качестве контроля ( n = 5–8). Иссекали икроножные мышцы обеих конечностей и взвешивали через 14 дней. Как сообщалось ранее (Bodine et al., 2001), эти мышцы в WT потеряли ~ 35% своей массы, но только 20% в MuRF1 — / — (P <0.001; неопубликованные данные). Мышцы икроножной мышцы мышей, экспрессирующих полноразмерный Myc-MuRF1, уменьшились в весе в той же степени, что и у WT (36% против 41%, соответственно; фиг. 1 E). Напротив, мышцы, экспрессирующие нефункциональный Myc-ΔR-MuRF1, уменьшились в весе всего на 22% (что напоминало уменьшение MuRF1 на 20% — / — ) по сравнению с 38% снижением у однопометников дикого типа (P < 0,01; рис. 1 E). Таким образом, домен безымянного пальца MuRF1 (то есть его функция как убиквитинлигаза) важен для быстрой атрофии после денервации, и потеря этого домена эквивалентна потере всего белка.
MuRF1, как сообщается, связан с миофибриллами (Centner et al., 2001), хотя иммунофлуоресцентное исследование сердечных миоцитов обнаружило MuRF1 также в цитоплазме (McElhinny et al., 2002). После выделения миофибрилл из икроножных мышц мышей MuRF1 myc / myc , ΔR-MuRF1 myc / myc и мышей WT, мы с удивлением обнаружили, что подавляющее большинство Myc-MuRF1 и Myc-ΔR -MuRF1 и эндогенный MuRF1 у мышей WT находились в цитозольной фракции (3000 г супернатанта) (рис.S3). Следовательно, если MuRF1 действительно связан с миофибриллами, это слабое, легко разрушаемое взаимодействие.
Для идентификации субстратов MuRF1, потерянных в результате денервации in vivo, мы иммунопреципитировали Myc-ΔR-MuRF1 из икроножных мышц (3000 г супернатанта ) из гомозиготного ΔR-MuRF1 тэг myc / myc и против его мышей, антител к Myc / myc промывали полученные иммунопреципитаты, как описано выше.Анализ белков, связанных с MuRF1, с помощью масс-спектрометрии идентифицировал тот же набор миофибриллярных белков, которые были осаждены с использованием иммобилизованного GST-MuRF1 (таблица I).
Чтобы прояснить роль MuRF1 в атрофии денервации и изучить, теряется ли какой-либо из этих саркомерных белков по-разному во время этого процесса, мы выделили миофибриллы из икроножных мышц, и после SDS-PAGE идентифицировали составляющие полосы белка с помощью окрашивания кумасси синим и масс-спектрометрии. (Рисунок.S4 А). Затем мы проанализировали с помощью SDS-PAGE равные количества белков из миофибриллярной фракции иннервируемых и денервированных мышц мышей MuRF1 myc / myc и ΔR-MuRF1 myc / myc и измерили интенсивность окрашенных специфических полос. с кумасси синим методом денситометрии (рис. S4 B). Масса мышц у животных ΔR-MuRF1 myc / myc и MuRF1 myc / myc уменьшилась аналогичным образом (14% [P <0,05] против 18% [P <0.001] соответственно) через 10 дней после денервации (неопубликованные данные). Несмотря на потерю белков, относительные концентрации подавляющего большинства миофибриллярных белков при 10-дневной денервации существенно не отличались от таковых в иннервируемых мышцах (рис. 3 A).
Неожиданно оказалось, что у животных, экспрессирующих Myc-MuRF1, относительные концентрации миофибриллярных MyLC2 и MyBP-C заметно снизились (на 47% и 28% соответственно; P <0.05) ниже уровня иннервируемых органов управления (рис. 3 A). Напротив, у мышей ΔR-MuRF1 myc / myc относительные количества этих регуляторных белков существенно не изменились после денервации (рис. 3А) (это уменьшение количества MyBP-C не связано с медленным быстрое изменение типа волокна, которое происходит после денервации [Weydert et al., 1987], потому что быстрая и медленная изоформы MyBP-C имеют одинаковую молекулярную массу и появляются в виде одной полосы на гелях, окрашенных синим кумасси). Эта дифференциальная потеря MyLC2 и MyBP-C заслуживает внимания, поскольку концентрации MyHC и актина в миофибриллах не изменились в денервированных мышцах обеих групп.
Поскольку MyLC1 и тропонин-I мигрируют одинаково при SDS-PAGE, их концентрации не могут быть измерены этим методом. Вместо этого мы проанализировали их уровни с помощью иммуноблоттинга (рис. 3 B). Подобно содержанию MyLC2 и MyBP-C, уровень MyLC1 был ниже в миофибриллярной фракции денервированной мышцы, чем в иннервируемой мышце мышей MuRF1 myc / myc , но не снижался в ΔR-MuRF1 myc / myc животных (рис.3 Б). Мы также подтвердили с помощью иммуноблоттинга, что относительные концентрации миофибриллярного MyHC и актина не различались в денервированных и иннервируемых мышцах в это время (рис. 3 B). Напротив, содержание тропонина-I, сердечная изоформа которого убиквитилирована MuRF1 и деградирует в культивируемых кардиомиоцитах (Kedar et al., 2004), не изменилось при денервации через 10 дней. Вместе эти данные показывают, что миофибриллы MyLC1, MyLC2 и MyBP-C преимущественно деградируют во время атрофии денервации по MuRF1-зависимому механизму, но другие потенциальные субстраты MuRF1 (например,g., компонент тонкой нити, тропонин-I) нет.
Чтобы подтвердить, что MyLC1, MyLC2 и MyBP-C являются прямыми субстратами для MuRF1, мы измерили их убиквитилирование с помощью рекомбинантного MuRF1 с использованием различных E2 и Вестерн-блоттинга (рис. 4). MuRF1 убиквитилировал чистый MyLC1 (рис. 4 A), MyLC2 (рис. 4 B) и MyBP-C (рис. 4 C), а также MyHC (рис. 4 D) с тремя протестированными E2, Ubch2, UbcH5, или Ubch23 / Uev1, все из которых, как было ранее показано, поддерживают убиквитилирование сердечного тропонина-I с помощью MuRF1 (Kim et al., 2007). Актин в основном моноубиквитилирован MuRF1 и UbcH5 (рис. S5 C), хотя in vivo эта цепь может быть удлинена другой убиквитин-лигазой, функционирующей как E4 (Koegl et al., 1999), или MuRF1 с другим E2 (как недавно предложено для мишеней комплекса, способствующего анафазе [Rodrigo-Brenni and Morgan, 2007]).
Соломон и Голдберг (1996) ранее показали, что, когда актин и миозин находятся в миофибриллах или даже в актомиозиновых комплексах, эти белки были довольно устойчивы к убиквитин-зависимому протеолизу, в отличие от свободного актина или миозина (Solomon and Goldberg, 1996).Чтобы узнать, может ли MuRF1 непосредственно убиквитилировать белки в миофибрилле или эта структура должна быть сначала диссоциирована, мы исследовали, являются ли MyLC1 и MyLC2, связанные в актомиозиновых комплексах, также субстратами для убиквитилирования с помощью MuRF1 (мы не смогли измерить убиквитилирование MyBP-C в актомиозине потому что его нет в препарате актомиозин). Мы сравнили убиквитилирование чистых белков и параллельно с препаратом актомиозина из мышц кролика (который в дополнение к актину, MyHC и MyLC может содержать небольшое количество ассоциированных белков, например.g., тропомиозин, тропонин, α-актинин). Аналогичные количества каждого из белков присутствовали в реакциях либо в виде чистого белка, либо в сочетании с актомиозином (скорректировано после того, как мы определили количества каждого белка путем окрашивания гелей кумасси синим). MuRF1 убиквитилировал MyLC1 и MyLC2 в актомиозиновом комплексе со скоростью, сравнимой со свободными белками со всеми тремя тестируемыми E2 (рис. 4, A и B). Напротив, MyHC может быть убиквитилирован только как чистый белок, но не в составе актомиозинового комплекса (рис.4 D).
Чтобы проверить, эффективно ли убиквитилированы MyLC1, MyLC2 и MyBP-C, когда они связаны с миофибриллами, мы односторонне денервировали икроножные мышцы мышей WT на 3 или 10 дней, приготовили миофибриллы из этих и контралатеральных нормальных мышц и сравнили показатели убиквитилирование белков в миофибриллах чистым MuRF1. Как наблюдалось у животных MuRF1 myc / myc , MyLC2 и MyBP-C были преимущественно потеряны (по сравнению с актином) из икроножной мышцы через 10 дней после денервации, тогда как относительная концентрация MyHC не изменилась (рис.5 А, слева). Кроме того, иммуноблоттинг выявил более низкие уровни MyLC1 в миофибрилле денервированной мышцы, чем в контроле (рис. 5А, справа).
Специфический анализ иммунопреципитации был разработан для выделения убиквитилированных белков и их анализа с помощью иммуноблоттинга. Чтобы оценить убиквитилирование белков в миофибриллах нормальных или денервированных мышц с помощью MuRF1, мы использовали убиквитин, меченный 6-His, Ubch2 и E1.После инкубации при 37 ° C добавляли 5 М мочевину для разборки миофибрилл и обеспечения эффективной очистки убиквитилированного гистом материала с помощью никелевой колонки. Когда изолированные миофибриллы от нормальных мышей инкубировали с MuRF1, никакого значительного убиквитилирования актина или MyHC с помощью иммуноблоттинга нельзя было продемонстрировать (неопубликованные данные). Напротив, MyLC1, MyLC2 и MyBP-C внутри миофибриллы были эффективно убиквитилированы как в контрольных, так и в денервированных мышцах (Fig. 5 B). Таким образом, в интактной миофибрилле и даже в актомиозиновом комплексе MyHC и актин (в отличие от MyLC1, MyLC2 и MyBP-C) проявляют лишь ограниченную чувствительность к убиквитилированию, т.е.е., намного меньше, чем очищенные MyHC и актин. Поскольку известно, что MyLC1, MyLC2 и MyBP-C стабилизируют толстые филаменты, их дифференциальное убиквитилирование и деградация может способствовать убиквитилированию, разборке и деградации других саркомерных компонентов.
Часто предлагалось, чтобы начальная стадия разборки, такая как протеолитическое расщепление кальпаинами (Goll et al., 1992) или каспазами (Du et al., 2005) может способствовать общей деградации миофибриллярного аппарата. Однако мы показали выше, что MyLC1, MyLC2 и MyBP-C преимущественно теряются во время атрофии денервации по MuRF1-зависимому механизму (рис. 3) и могут убиквитилировать с помощью MuRF1, когда все еще связаны с миофибриллами (рис. 5 B). Кроме того, очищенный растворимый актин и MyHC убиквитилируются MuRF1 более эффективно, чем когда эти белки связаны с другими компонентами миофибриллы или даже в актомиозиновом комплексе (рис.4). Эти находки повышают вероятность того, что потеря MyLC1, MyLC2 и MyBP-C может увеличивать восприимчивость других миофибриллярных компонентов к MuRF1-зависимой деградации.
Чтобы проверить эту гипотезу, мы измерили содержание компонентов толстой и тонкой нити через 14 дней после денервации. Контрольные и денервированные мышцы мышей MuRF1 myc / myc и ΔR-MuRF1 myc / myc гомогенизировали, и изолированные миофибриллы анализировали с помощью SDS-PAGE и окрашивания кумасси синим.Измерение интенсивности конкретных полос выявило дифференциальную потерю MyHC из миофибрилл на 14-й день денервации у мышей MuRF1 myc / myc (рис. 6 A), которая не была очевидна через 10 дней после денервации (рис. 3 и неопубликованные данные). ). Напротив, количество MyHC и ни одного из компонентов толстых филаментов (т.е. MyLC1, MyLC2 и MyBP-C) уменьшилось у ΔR-MuRF1 myc / myc животных. Эта более медленная потеря MyHC согласуется с гипотезой о том, что начальная селективная деградация стабилизирующих толстые филаменты белков MyLC1, MyLC2 и MyBP-C увеличивает доступность MyHC для убиквитилирования с помощью MuRF1.
В отличие от этих компонентов толстых волокон, в это время после денервации компоненты тонких волокон, актин, тропомиозин и α-актинин были потеряны из миофибрилл аналогичным образом в MuRF1 myc / myc и ΔR-MuRF1 myc / myc животных (рис. 6 А). Таким образом, хотя MuRF1 необходим для деградации и разборки толстых филаментов во время атрофии денервации, тонкие филаменты деградируют по особому механизму, который не требует MuRF1 или MuRF1-зависимого убиктиилирования других компонентов, но может включать другую убиквитинлигазу.
Настоящие результаты проясняют критическую роль MuRF1 в мышечной атрофии. Хотя другие убиквитинлигазы также индуцируются во время атрофии (например, атрогин-1 [Gomes et al., 2001], который также важен для быстрой атрофии [Bodine et al., 2001], а также E3α-II [Kwak et al. ., 2004]), MuRF1 необходим для большей части увеличения конъюгатов убиквитина во время денервационной атрофии (Рис. 2).Таким образом, MuRF1 либо убиквитилирует большинство белков, которые разрушаются через 10 дней после денервации, либо его действия позволяют действовать другим лигазам. В любом случае MuRF1 явно является очень общей лигазой, влияющей на многие сократительные (таблица I), растворимые и ядерные белки (неопубликованные данные), что также предполагалось в двухгибридных исследованиях (Witt et al., 2005; Hirner et al. , 2008). Более того, наши исследования предполагают участие убиквитилирования MuRF1 в селективной потере MyBP-C, MyLC1 и MyLC2, а также в более медленной деградации MyHC и, таким образом, в разборке и деградации миофибриллярных белков, что является определяющим признаком атрофии процесс.
Инактивация способности MuRF1 связывать E2 путем делеции его мотива безымянного пальца ослабляла атрофию и предотвращала дифференциальную потерю MyBP-C, MyLC1 и MyLC2 из миофибрилл (рис. 3). Их избирательная потеря является новым признаком атрофии мышц и, по-видимому, имеет регуляторное значение. Поскольку эти белки, по-видимому, имеют решающее значение для стабилизации миофибрилл (см. Ниже), снижение потери MyHC у мышей ΔR-MuRF1 myc / myc может быть прямым следствием сохранения MyLC1, MyLC2 и MyBP- С.Также удивительными были открытия, что эти компоненты толстых филаментов разлагаются быстрее и по отдельному механизму от компонентов тонких филаментов, актина, тропомиозина и α-актинина, деградация которых не зависит от MuRF1.
Поскольку MuRF1 необходим для быстрой атрофии, но не для нормального роста или функции мышц, идентификация его субстратов важна для понимания механизмов потери белка.Мы идентифицировали ряд миофибриллярных компонентов в качестве новых субстратов для MuRF1 in vivo, все из которых важны для целостности мышц и сократительной активности. Мутации в каждом из них вызывают серьезные нарушения, характеризующиеся нарушением порядка миофибрилл и нарушением сократительной функции (Clark et al., 2002). Хотя ранее сообщалось, что сердечный тропонин-I является субстратом MuRF1 (Kedar et al., 2004), тропонин-I не может быть иммунопреципитирован с помощью MuRF1 из этих скелетных мышц и не теряется с помощью MuRF1-зависимого механизма после денервации.MyHC накапливается у мышей, лишенных MuRF1 и MuRF3 (Fielitz et al., 2007), и разрушается MuRF1-зависимым образом при обработке эмбриональных мышечных трубок дексаметазоном (Clarke et al., 2007). Соответственно, снижение содержания MyHC по MuRF1-зависимому механизму было ясно видно на 14-й день денервации (фиг. 6). Однако в миофибриллах иннервируемых мышц MyHC не убиквитилируется с помощью MuRF1 и не является предпочтительным субстратом MuRF1 даже через 10 дней после денервации (Fig. 3).
Примечательно, что эти различные миофибриллярные белки могли быть преципитированы с помощью GST-MuRF1 из цитозольной фракции и были убиквитилированы чистым MuRF1.Растворимые пулы этих миофибриллярных компонентов описаны у кур (Horvath and Gaetjens, 1972) и были обнаружены в экстрактах мышц мышей (неопубликованные данные). Возможно, эти растворимые компоненты функционируют либо как предшественники зрелого сократительного аппарата, либо как компоненты, высвобождаемые во время миофибриллярного обмена, или могут представлять собой фракцию миофибриллярных белков, которые легко высвобождаются при гомогенизации (Etlinger et al., 1975). Однако эти белки не накапливались в цитозоле при атрофии, когда они были потеряны из миофибриллы (неопубликованные данные).
Хотя давно было признано, что основные миофибриллярные белки разлагаются убиквитин-протеасомным путем во время атрофии (Solomon and Goldberg, 1996), идентификация MuRF1 как критической лигазы, которая действует на миофибриллярный компартмент (возможно, вместе с другими E3) ) предполагает роль убиквитилирования в разборке миофибрилл. Примечательно, что очищенный мономерный актин и MyHC были эффективно убиквитилированы MuRF1, но не тогда, когда были связаны друг с другом в актомиозиновом комплексе или в интактной миофибрилле (рис.4, неопубликованные данные). Ранее Соломон и Голдберг (1996) продемонстрировали на грубых гомогенатах, что убиквитин-протеасомная система быстро разрушает мономерный актин и миозин, но этот процесс не был продемонстрирован в экстрактах, когда эти белки были связаны друг с другом в актомиозиновом комплексе или миофибрилле. . Эти ранние открытия предполагают, что во время атрофии может существовать механизм высвобождения этих компонентов из миофибрилл или повышения чувствительности актина, миозина и других миофибриллярных белков к убиквитилированию.Однако, основываясь на настоящих выводах, больше нет необходимости постулировать такую систему выпуска (см. Ниже).
MuRF1 может убиквитилировать эти субстраты вместе с Ubch2, который образует полиубиквитиновые цепи, связанные с K-48, и Ubch23 / Uev1, который образует цепочки, связанные с K-63, и UbcH5, который образует смешанные цепи (Kim et al., 2007) (Рис.4). Однако неясно, какие E2s функционируют в протеолизе с помощью MuRF1 или какие типы цепей убиквитина они образуют в нормальных или атрофированных мышцах.Хотя K-63-связанные цепи убиквитина, как полагают, выполняют функции, отличные от протеасомной деградации in vivo, недавние открытия показывают, что цепи K63 могут нацеливать субстраты на протеасомы in vitro и in vivo (Saeki et al., 2009). Кроме того, природа и содержание E2 могут влиять на скорость деградации белка и, следовательно, на скорость атрофии.
Особенно интересно то, что MyLC1, MyLC2 и MyBP-C избирательно разлагаются MuRF1-зависимым образом после денервации (рис.3), и что эти белки, в отличие от MyHC и актина, эффективно убиквитилируются с помощью MuRF1 внутри миофибриллы (Fig. 5B, неопубликованные данные). Таким образом, эти критически важные стабилизирующие белки могут быть субстратами внутри миофибриллы без диссоциации или протеолитического процессинга. Более того, восприимчивость MyHC к убиквитилированию и деградации увеличивается через 14 дней после денервации (рис. 6 A), предположительно из-за того, что уровни MyBP-C и MyLC в миофибриллах снижены. Соответственно, миозин может быть извлечен из сердечной толстой нити только после снижения содержания MyBP-C на 20% (Куликовская и др., 2007). Напротив, компоненты тонких филаментов, актин, тропомиозин и α-актинин, были потеряны из миофибрилл через 14 дней после денервации по механизму, который не требует MuRF1 или MuRF1-зависимой потери MyBP-C и MyLC. Однако также возможно, что MuRF1 может нацеливаться на белки деградации, связанные с решеткой Z-диска, потому что филамин C был осажден вместе с MuRF1 (Таблица I). Альтернативно, другой E3, индуцированный при атрофии, может способствовать разборке Z-диска или тонких нитей и функционировать у животных ΔR-MuRF1.
Хотя эти методы не продемонстрировали снижения абсолютных уровней сократительных белков до 10 дней после денервации, их деградация должна была ускориться намного раньше, потому что потеря белков является результатом интегрированных эффектов усиленного протеолиза с течением времени. Любые небольшие изменения в содержании миофибрилл (например, уменьшение на 10–20%), которые происходят раньше после денервации, вероятно, не могут быть обнаружены методами, используемыми здесь.Соответственно, наше открытие, что MuRF1 индуцируется через 3 дня после денервации (Fig. 5 A), означает, что MuRF1 функционирует максимально рано при атрофии (и до того, как будет обнаружена какая-либо потеря миофибриллярных белков). MuRF1 может способствовать деградации растворимых цитозольных компонентов, многие из которых связаны с MuRF1 (неопубликованные данные).
Примечательно, что снижение мышечной массы может быть продемонстрировано в течение первой недели после денервации (Furuno et al., 1990) и, по-видимому, не связаны с MuRF1-зависимой потерей миофибриллярных компонентов (Fig. 6 A). Однако другие E3 быстро индуцируются во время атрофии (например, атрогин-1) и могут способствовать ускорению деградации. Кроме того, недавние исследования показали, что аутофагия быстро активируется во время денервации-атрофии и, по-видимому, способствует разрушению митохондрий и цитозольных белков, возможно, в течение первой недели после денервации (Mammucari et al., 2007; Zhao et al., 2007; O ‘ Leary and Hood, 2009), прежде чем можно будет обнаружить какую-либо потерю миофибриллярного миозина и актина.
Т.о., во время атрофии, MuRF1, по-видимому, функционирует на начальных этапах разборки толстых филаментов, по-видимому, путем выборочного убиквитилирования определенных ключевых регуляторных компонентов, чья деградация должна способствовать разрушению оставшихся компонентов толстых филаментов. Экстракция этих убиквитилированных регуляторных белков из миофибриллы может быть напрямую связана с их деградацией протеасомой 26S, 19S компонент которой катализирует АТФ-зависимое разворачивание и транслокацию субстратов в ядро протеасомы 20S (Smith et al., 2005). Альтернативно, АТФ-зависимая экстракция этих белков может катализироваться комплексом p97 / VCP, который извлекает убиквитин-конъюгированные белки из мембран ER в связанном с ER пути деградации (Ye et al., 2001) и играет важную роль в миофибриллах. сборки (Janiesch et al., 2007). Дополнительным доказательством критической регуляторной роли миофибриллярного MyBP-C является то, что мРНК этого белка также избирательно снижается во время атрофии денервации (неопубликованные данные).
MyBP-C и MyLC2 являются критическими модуляторами мышечной сократимости, и их потеря может быть причиной некоторых изменений сократительных свойств, наблюдаемых после денервации (Zorzato et al., 1989; Trachez et al., 1990). Экстракция MyBP-C из ободранных волокон приводит к увеличению чувствительности к развитию силы Ca 2+ (Hofmann et al., 1991). Удаление миофибриллярного MyLC2 оказывает такое же влияние на кривую удлинения Ca 2+ , что и удаление MyBP-C. Оба белка, таким образом, по-видимому, уменьшают несоответствующие сокращения за счет уменьшения вероятности связывания миозиновых головок с актином (Moss et al., 1983). Кроме того, в сердце увеличение содержания MyBP-C усиливает АТФазную активность актомиозина, но не в случае удаления MyLC2, и этот эффект снова проявляется при восстановлении MyLC2 (Margossian, 1985).
Через несколько дней после денервации сократительные свойства мышцы изменяются (т.е. происходит удлинение периодов сокращения и расслабления), по-видимому, из-за изменений внутриклеточных концентраций Ca 2+ (Zorzato et al., 1989), а также повышенная чувствительность миофибрилл к Ca 2+ (Trachez et al., 1990). Как отмечалось выше, экстракция MyBP-C или MyLC2 также приводит к повышенной чувствительности миофибрилл к Ca 2+ .Поскольку Ca 2+ менее эффективно выкачивается из цитозоля денервированных мышц (Finol et al., 1981), избирательная потеря этих регуляторных компонентов может вносить вклад в их измененные сократительные свойства. Еще одна характеристика атрофированной мышцы — прогрессирующее снижение силы сокращений. Поскольку MyLC1 необходим для производства полной силы мышечным миозином (VanBuren et al., 1994), его избирательная потеря (а также последующая потеря MyHC) должна способствовать потере напряжения после денервации.
Хотя убиквитилирование миофибриллы и избирательная деградация критических стабилизирующих белков MyBP-C, MyLC1 и MyLC2 под действием MuRF1 является новым привлекательным механизмом потери толстых филаментов, еще предстоит доказать, что эти последовательные этапы важны для потеря MyHC и быстрая атрофия. Также будет важно установить характер последующих шагов, которые приводят к потере тонких нитей во время атрофии, а также во время более медленного оборота миофибриллярных компонентов в нормальной мышце.Этот механизм (убиквитилирование ключевых регуляторных компонентов в миофибрилле) сильно отличается от частого предположения, что начальная атака на сократительный аппарат осуществляется эндопротеазами, такими как каспазы (Du et al., 2004) или кальпаины (Tidball and Spencer, 2002). ). Хотя такие механизмы могут функционировать в определенных условиях апоптоза (Du et al., 2004), прямые доказательства того, что эти протеазы играют существенную или основную роль, отсутствуют, и настоящие результаты демонстрируют совершенно другой MuRF1-зависимый механизм, который, по-видимому, функционирует in vivo. на уровне миофибриллы.
Как остановить атрофию мышц после инсульта: 3 полезных совета
Атрофия мышц после инсульта вызывает уменьшение мышечной массы. Это приводит к снижению силы и повышенному риску травм.
Многие пациенты, перенесшие инсульт, испытывают это состояние во время реабилитации, и оно может быть серьезным. К счастью, есть надежда на выздоровление.
В этой статье вы узнаете 3 основные причины атрофия мышц после инсульта и как обратить ее последствия.
Причины атрофии мышц после инсульта
Мышечная атрофия — это ухудшение мышечной ткани из-за периоды бездействия.
Все мышцы истощаются, если вы их не тренируете регулярно. Например, человек, который носит повязку на руке в течение нескольких недель. потеряет мышечную массу на этой руке. Это касается и после инсульта.
Есть несколько причин, по которым вы можете испытывать мышечную атрофия после инсульта. Три самых распространенных:
- Выявлено, что не используется. Инсульт часто вызывает слабость или паралич одной стороны тела. Если ваша правая рука слаба, вы можете начать полагаться на левую руку при выполнении большинства задач. Это может привести к тому, что вы научитесь не использовать — состоянию, при котором ваш мозг теряет связь с вашими мышцами. Это, в свою очередь, приводит к атрофии мышц.
- Длительная госпитализация. Если инсульт был тяжелым, пациенту, возможно, придется оставаться в больнице в течение нескольких недель. Это продолжительное бездействие приведет к разрушению мышц.Вот почему так важно принять участие в терапии, как только она будет предложена вам в больнице.
- Недоедание. Наконец, инсульт может вызвать трудности с пережевыванием и глотанием пищи (также известные как дисфагия). Это может привести к недоеданию, которое в сочетании с бездействием может ускорить атрофию мышц.
Как видите, инсульт вызывает ряд факторов, которые могут привести к потере мышечной массы. Поэтому, чтобы обратить вспять атрофию мышц, вам необходимо: устраните эти основные причины.
Профилактика и лечение мышечной атрофии
Последствия атрофии мышц серьезны, так как она создает порочный круг. Чем сильнее ухудшаются ваши мышцы, тем меньше у вас сил двигать мышцами. И чем меньше вы двигаете мышцами, тем больше у вас мышц атрофируется.
Вот почему по возможности важно принять меры, чтобы как можно скорее предотвратить атрофию мышц. Однако даже если ваша мышечная масса уже уменьшился, эти меры также могут помочь вам обратить вспять атрофию и предотвратить ухудшение.
Ниже приведены несколько лучших способов предотвращения и лечить атрофию мышц после инсульта:
1. Лечите гемиплегию пассивными упражнениями
Если вы боретесь с гемиплегией, то есть параличом с одной стороны тела — вам крайне важно немедленно решить эту проблему. Чем дольше ты не двигайте пораженной стороной, тем больше вы потеряете мышечной массы.
К счастью, даже если вы сейчас не можете двигаться самостоятельно, есть способы обратить вспять последствия инсульта.
Вы начнете с пассивных упражнений, которые выполняет терапевт, который двигает за вас пораженные конечности. Когда ваш терапевт двигает вашими конечностями, это стимулирует ваш мозг и активирует процесс, известный как нейропластичность.
Под нейропластичностью понимается способность мозга перестраивать нервную систему. пути. Это позволяет неповрежденным участкам мозга брать на себя функции из поврежденных участков. Вы можете активировать нейропластичность с помощью повторяющихся и даже пассивное движение.
Следовательно, чем больше вы пассивно двигаете руками или ногами, тем больше вы будете стимулировать свой мозг и формировать новые нейронные пути между ваш мозг и мышцы.В конце концов, вы должны восстановить небольшое количество движение.
Когда это произойдет, вы можете перейти к следующему шагу.
2. Укрепление мышц с помощью активных упражнений
Когда вы восстановите достаточное количество движений в пораженных мышцах, вы можете приступить к выполнению упражнений по активной реабилитации после удара.
Чем больше вы практикуетесь, тем больше вы укрепляете нервные пути, которые помогают вам двигаться. В конце концов, ваша сила и контроль над ваши мышцы улучшатся.
Вы можете начать с простого движения пораженной рукой или ногой. без сопротивления.Но по мере того, как ваши способности улучшаются, вы должны продвигаться к большему. сложные виды деятельности, такие как упражнения с отягощением. Это будет стимулировать рост мышц и обратная атрофия.
Однако для полного восстановления мышечной силы потребуется большое количество повторений. Например, исследования на животных показали, что требуется от 400 до 600 повторений сложных функциональных задач в день, чтобы вызвать нейропластичность и восстановить движения.
Это может быть трудно сделать самостоятельно, но многие пациенты с инсультом обнаруживают, что инструменты реабилитации, такие как домашняя терапия FitMi от Flint Rehab, мотивируют их выполнять больше повторений.Фактически, средний пациент выполняет примерно в 23 раза больше повторений с FitMi, чем с традиционной терапией.
Чем больше упражнений вы выполните, тем быстрее вы измените свое атрофия мышц после инсульта и восстановление функции.
3. Профилактика недоедания с помощью логопедических упражнений
Наконец, важно бороться с дисфагией после инсульта, чтобы предотвратить истощение и атрофию мышц.
Лучше всего это делать с помощью логопедических упражнений.Как и другие упражнения на инсульт, логопедические упражнения используют силу нейропластичности вашего мозга. Это поможет вам восстановить контроль над глотательными и жевательными мышцами.
Если вы сможете вылечить дисфагию на раннем этапе выздоровления, вы сможете уберечь себя от истощения. Это, в свою очередь, предотвратит атрофию мышц.
Восстановление атрофии мышц после инсульта
Таким образом, мышечная атрофия возникает, когда мышцы ухудшаются. из-за малоподвижности и неправильного питания.
Это запускает порочный круг, в котором мышцы ослабевают и становятся все более запущенными, что приводит к еще большей атрофии.
Чтобы разорвать этот цикл и обратить вспять атрофию мышц после инсульта, все, что вам нужно сделать, это двигаться. Если это невозможно, вы можете начать с пассивного упражнения и постепенно переходите к активной нагрузке на вес тела.
Атрофия мышц и ее различные причины
Атрофия скелетных мышц относится к потере мышечной массы. Истощенные мышцы выглядят сморщенными и меньшими по размеру по сравнению с непораженной стороной.Также будет соответствующая потеря мышечного тонуса и силы. Мышечные отходы из-за неиспользования, отека, повреждения нервов или болезненного процесса. Если не устранить причину, это может привести к серьезному снижению качества жизни. Там, где нет необратимых повреждений, упражнения или физическая активность — лучший способ предотвратить истощение мышц.
Причины атрофии мышц
НеиспользованиеРаспространенной причиной атрофии мышц является неиспользование. Это происходит, когда мышцы недостаточно напряжены из-за недостатка физической активности или из-за боли.Это обычно наблюдается у людей, ведущих малоподвижный образ жизни, и пожилых людей с пониженным уровнем активности. Было замечено, что мышцы начинают истощаться в течение 4 часов после начала постельного режима.
ОтекОтек суставов или выпот также могут вызвать атрофию мышц и потерю их силы. Это особенно заметно при травме колена. Например, разрыв ПКС, при котором отек препятствует активации мышцы бедра. Постепенно заставляя мышцы терять свою функцию.Следовательно, лечение отека является одним из наиболее важных аспектов острой спортивной травмы.
Повреждение нерваНейрогенная атрофия мышц возникает при повреждении нерва, нарушающем его нормальную функцию. Например, нерв, затронутый протрузией диска со смещением дискора, не сможет активировать свои соединительные мышцы. Мышцы, которые не активируются в течение длительного периода времени, тратят впустую. Это может стать серьезной проблемой, так как пациент может в конечном итоге потерять контроль над своими конечностями или испытать боль и дискомфорт из-за сморщенных, ослабленных мышц.
БолезньКахексия, или «синдром истощения тела», и мышечная дистрофия являются примерами заболеваний, которые вызывают прогрессирующее истощение мышц. Кахексия часто наблюдается у пациентов со СПИДом, раком и другими серьезными хроническими заболеваниями. У этой группы пациентов могут возникнуть трудности с восстановлением мышечной ткани. Популярный сингапурский телевизионный актер Чу Чор Менг — местный пример, у которого была диагностирована мышечная дистрофия.
Упражнения — самый важный элемент в восстановлении мышечной ткани.Чтобы накачать мышцы, упражнения также можно дополнить специальной диетой. Если упражнения нецелесообразны, особенно для пациентов, прикованных к постели, электрическая стимуляция нерва атрофированной области может помочь уменьшить тяжесть атрофии мышц за счет искусственной активации мышц пациента.
Атрофия мышц может быть серьезной и изнурительной, приводя к снижению качества вашей жизни. Если атрофия мышц появляется внезапно и без видимой причины, рекомендуется обратиться к врачу.