Синдром кошачьего глаза у человека фото при смерти: Американка с синдромом кошачьего глаза стала моделью: События: Из жизни: Lenta.ru
Принт | Бизнес как он есть
Налоговый кодекс земельный налог
29.05.2020
Комментариев нет
НАЛОГОВЫЙ КОДЕКС — Глава 31. ЗЕМЕЛЬНЫЙ НАЛОГ Статья 387. Общие положения1. Земельный налог (далее в настоящей главе — налог) устанавливается […]
Читать далее →
Аренда автомобиля проводки
29.05.2020
Комментариев нет
Бухгалтерский и налоговый учет аренды автомобиля в 2020 году Любой вид бизнеса не может существовать без автотранспорта, так как это […]
Читать далее →
Договор аренды с экипажем
29.05.2020
Комментариев нет
Как правильно составить договор аренды автомобиля с водителем, образец документа Юридическая консультация > Административное право > Оформление документов > Как […]
Работа ППС
29. 05.2020
05.2020
Комментариев нет
Как устроиться в ППС без армии Довольно часто молодежь, не служившая ранее в вооружённых силах нашей страны, задаётся вопросом: как […]
Читать далее →
Код отчетного периода
29.05.2020
Комментариев нет
Коды в декларации по налогу на прибыль Актуально на: 19 марта 2019 г. Часть информации на титульном листе декларации по […]
Читать далее →
Рассчитать пособие по уходу
29.05.2020
Комментариев нет
Онлайн калькулятор пособия по уходу за ребенком до 1.5 лет для расчета в 2020 году Выше представлен онлайн калькулятор, в […]
Читать далее →Расчетный лист по зарплате расшифровка кодов
29. 05.2020
05.2020
Комментариев нет
Как заставить дирекцию выдавать ежемесячные корешки по зарплате? Добрый вечер! Какой-то отдельной программы для этого нет. Даже самая простая 1С: […]
Читать далее →
Профстандарт уборщика
28.05.2020
Комментариев нет
Профстандарт уборщика служебных помещений Профессиональный стандарт уборщика служебных помещений в государственном секторе носит обязательный характер, а в бизнес-сфере его используют […]
Читать далее →
Выговор в трудовую книжку
28.05.2020
Комментариев нет
Заносится ли выговор в трудовую книжку? Обновление: 16 декабря 2016 г. У начинающих кадровиков нередко возникает вопрос, заносится ли в […]
Читать далее →
КПП, что это?
28. 05.2020
05.2020
Комментариев нет
Что такое КПП банка и как его узнать? Сегодня у многих людей возникает необходимость оформления определенных финансовых документов, а также […]
Читать далее →
Принт | Бизнес как он есть
Налоговый кодекс земельный налог
29.05.2020
Комментариев нет
НАЛОГОВЫЙ КОДЕКС — Глава 31. ЗЕМЕЛЬНЫЙ НАЛОГ Статья 387. Общие положения1. Земельный налог (далее в настоящей главе — налог) устанавливается […]
Читать далее →
Аренда автомобиля проводки
29.05.2020
Комментариев нет
Бухгалтерский и налоговый учет аренды автомобиля в 2020 году Любой вид бизнеса не может существовать без автотранспорта, так как это […]
Читать далее →
Договор аренды с экипажем
29. 05.2020
05.2020
Комментариев нет
Как правильно составить договор аренды автомобиля с водителем, образец документа Юридическая консультация > Административное право > Оформление документов > Как […]
Читать далее →
Работа ППС
29.05.2020
Комментариев нет
Как устроиться в ППС без армии Довольно часто молодежь, не служившая ранее в вооружённых силах нашей страны, задаётся вопросом: как […]
Читать далее →
Код отчетного периода
29.05.2020
Комментариев нет
Коды в декларации по налогу на прибыль Актуально на: 19 марта 2019 г. Часть информации на титульном листе декларации по […]
Читать далее →
Рассчитать пособие по уходу
29. 05.2020
05.2020
Комментариев нет
Онлайн калькулятор пособия по уходу за ребенком до 1.5 лет для расчета в 2020 году Выше представлен онлайн калькулятор, в […]
Читать далее →
Расчетный лист по зарплате расшифровка кодов
29.05.2020Комментариев нет
Как заставить дирекцию выдавать ежемесячные корешки по зарплате? Добрый вечер! Какой-то отдельной программы для этого нет. Даже самая простая 1С: […]
Читать далее →
Профстандарт уборщика
28.05.2020
Комментариев нет
Профстандарт уборщика служебных помещений Профессиональный стандарт уборщика служебных помещений в государственном секторе носит обязательный характер, а в бизнес-сфере его используют […]
Читать далее →
Выговор в трудовую книжку
28. 05.2020
05.2020
Комментариев нет
Заносится ли выговор в трудовую книжку? Обновление: 16 декабря 2016 г. У начинающих кадровиков нередко возникает вопрос, заносится ли в […]
Читать далее →
КПП, что это?
28.05.2020
Комментариев нет
Что такое КПП банка и как его узнать? Сегодня у многих людей возникает необходимость оформления определенных финансовых документов, а также […]
Читать далее →
Трипликация 600 т.п.н. в критической области синдрома кошачьего глаза вызывает аноректальные, почечные и преаурикулярные аномалии в семье из трех поколений
Введение
Синдром кошачьего глаза (CES) имеет широкий спектр клинических признаков. Наиболее часто выявляемыми клиническими признаками у пациентов с диагнозом CES являются преаурикулярные метки или ямки (81–87%), аноректальные пороки развития (атрезия, переднее расположение) (73–81%), урогенитальные пороки развития (например, одностороннее отсутствие почки) (71%). , колобома глаза (55–61%) и врожденные аномалии сердца (например, аномальный возврат легочных вен, тетрада Фалло, персистенция левой верхней полой вены, аномалии клапана) (50–63%), 1 , но ни один из них не обязательно присутствует. 2, 3 Психическое развитие в целом нормальное или почти нормальное 2, 3, 4, 5 и многие другие аномалии описаны у меньшего числа пациентов. Только у 41% пациентов выявляются все три основные видимые аномалии (колобома, анальные аномалии и преаурикулярные аномалии), что может свидетельствовать о недостаточной диагностике СЭС. 2, 3
, колобома глаза (55–61%) и врожденные аномалии сердца (например, аномальный возврат легочных вен, тетрада Фалло, персистенция левой верхней полой вены, аномалии клапана) (50–63%), 1 , но ни один из них не обязательно присутствует. 2, 3 Психическое развитие в целом нормальное или почти нормальное 2, 3, 4, 5 и многие другие аномалии описаны у меньшего числа пациентов. Только у 41% пациентов выявляются все три основные видимые аномалии (колобома, анальные аномалии и преаурикулярные аномалии), что может свидетельствовать о недостаточной диагностике СЭС. 2, 3
CES вызывается дупликацией или тройничеством части хромосомы 22. В большинстве случаев обнаруживается бисателлитная малая дицентрическая сверхштатная хромосома, состоящая из дважды хромосомы 22pter до проксимального фрагмента 22q11.21, цитогенетически записываемая как inv dup(22)(q11.21). 4, 5 Внутрихромосомная дупликация или тройная дупликация проксимального фрагмента 22q также может давать сходный фенотип с частичным или полным спектром клинических характеристик. Не наблюдается существенной разницы в тяжести синдрома между пациентами с вариантом частичной трисомии, возникающей в результате дупликации, или вариантом частичной тетрасомии, возникающей в результате тройной. 6, 7, 8, 9
Не наблюдается существенной разницы в тяжести синдрома между пациентами с вариантом частичной трисомии, возникающей в результате дупликации, или вариантом частичной тетрасомии, возникающей в результате тройной. 6, 7, 8, 9
Несколько раз сообщалось о мозаицизме нештатной хромосомы inv dup(22), но степень мозаицизма не могла быть коррелирована с тяжестью фенотипа. 2 Также было опубликовано наследование нештатной хромосомы. 5, 10 В некоторых из этих случаев члены семьи имеют общие фенотипические характеристики, но в других случаях члены семьи демонстрируют фенотипическую изменчивость в диапазоне от более легких до более тяжелых фенотипов. Следовательно, трудно предсказать риск рецидива и фенотипическую тяжесть для потомства пациента с CES. Может существовать значительное совпадение между CES и другими синдромами с анальными, аурикулярными, сердечными и урогенитальными аномалиями, такими как синдром Таунса-Брокса (анальные, аурикулярные, сердечные и урогенитальные аномалии), вызванный мутациями в SALL1 , окуло-аурикуловертебральный спектр/синдром Гольденхара (офтальмологические, аурикулярные, сердечные и почечные аномалии) или ассоциация VACTERL (анальные, сердечные и почечные аномалии). Эти синдромы также очень изменчивы. 11
Эти синдромы также очень изменчивы. 11
В этом исследовании мы сообщаем о SALL1 — отрицательной мутации, семье из трех поколений, в которой четыре члена несут внутрихромосомную частичную трипликацию области CES длиной 600 т.п.н., вызывающую частичную тетрасомию критической области CES. Усиливаемая область меньше принятой в настоящее время критической области. В этом семействе существуют фенотипические вариации, но все члены-носители обнаруживают фенотипическое совпадение с CES. Обсуждается влияние амплифицированных и неамплифицированных генов критической области CES на фенотип у зарегистрированных пациентов. Представленные данные могут способствовать пониманию геноспецифической причинности особенностей CES.
Материалы и методы
Клиническая оценка пациента и семьи
Основной пациент — новорожденная девочка с вестибулярным анусом и преаурикулярной ушной ямкой слева. И мать, и бабушка по материнской линии перенесли операцию по поводу анальной атрезии и преаурикулярных ушных меток. Последний был также обнаружен у брата матери, который был известен и лечился от идиопатического дефицита гормона роста и у которого была односторонняя агенезия почки. Брат бабушки был известен с синдромом «Гольденхара» с глухотой, лицевой асимметрией, микротией, отсутствием одного слухового прохода и был описан как «простой». Другой брат умер новорожденным и, вероятно, также имел анальную аномалию (рис. 1).
Последний был также обнаружен у брата матери, который был известен и лечился от идиопатического дефицита гормона роста и у которого была односторонняя агенезия почки. Брат бабушки был известен с синдромом «Гольденхара» с глухотой, лицевой асимметрией, микротией, отсутствием одного слухового прохода и был описан как «простой». Другой брат умер новорожденным и, вероятно, также имел анальную аномалию (рис. 1).
Семейная родословная, стрелка указывает индекс пациента. Носители трипликации 22q11.1q11.21 обозначены звездочкой (*). Непроверенные члены семьи отмечены знаком вопроса, а «N» означает «не носитель» трипликации. Лица с незакрашенным символом не имеют фенотипических характеристик CES. Черный верхний правый квадрант обозначает порок развития заднего прохода, черный верхний левый квадрант обозначает преаурикулярные аномалии, черный нижний правый квадрант обозначает почечные аномалии, а черный нижний левый квадрант обозначает глухоту. У дяди пациента-индекса также был идиопатический дефицит гормона роста; на этом рисунке это не указано.
У дяди пациента-индекса также был идиопатический дефицит гормона роста; на этом рисунке это не указано.
Изображение в полный размер
Ни у одного из пострадавших не было врожденных аномалий сердца или глазных аномалий. Пробанду, двум ее братьям, матери и бабушке по материнской линии было проведено офтальмологическое обследование. Дедуля с синдромом Гольденхара не был доступен для обследования.
Цитогенетические и молекулярные оценки
Хромосомный анализ с использованием полос GTG был выполнен на стимулированных культурах периферической крови основного пациента с использованием стандартных методов. Выделение ДНК из клеток периферической крови исследуемых членов семьи проводили по стандартной методике. MLPA выполняли с использованием имеющегося в продаже набора SALSA MLPA P250-B1 (MRC-Holland, Амстердам, Нидерланды), содержащего зонды, специфичные к геномным областям, которые наиболее вероятно вовлечены в велокардиофациальный синдром, синдром Ди Джорджи, синдром дупликации 22q11. 2, дистальный 22q11. .2 синдром делеции, синдром делеции 22q13.3 и CES. Эксперименты MLPA проводились в соответствии с инструкциями производителя.
2, дистальный 22q11. .2 синдром делеции, синдром делеции 22q13.3 и CES. Эксперименты MLPA проводились в соответствии с инструкциями производителя.
FISH-анализ области CES был выполнен у основного пациента с клонами BAC RP11-155N18 и RP11-958h30, расположенными в 22q11.1 и 22q11.1q11.21, соответственно, и вместе покрывающими около 470 т.п.н. критической области CES. , 12, 13 , включая гены CECR1 , CECR2 , CECR4 , CECR5 , CECR6 и IL17RA . Эксперименты FISH проводили по стандартным методикам 14 с альфоидным зондом p19.0,22 в качестве контрольного зонда для центромеры 22. 15
Affymetrix Genome-Wide Human SNP Array 6.0 (Affymetrix, Санта-Клара, Калифорния, США) был проведен у основного пациента в соответствии с протоколами производителя. Анализ данных проводили с использованием программных пакетов Affymetrix Genotyping Console (GTC) и Chromosome Analysis Suite (ChAS). Результат был сравнен in silico с объединенными данными 94 эталонных образцов, состоящих из нормальных контрольных образцов, созданных собственными силами.
Результаты
Основываясь на клиническом подозрении на возможную перестройку области CES, скрининг MLPA у основного пациента показал частичное усиление этой области 22q11. Три из пяти зондов, специфичных для этой области (т. е. SLC25A18, BID и MICAL3 ), продемонстрировали усиление соотношения сигналов, предполагая, что число копий равно четырем. Самый проксимальный датчик для области CES в наборе ( IL17RA ) показал нормальное соотношение, как и самый дистальный датчик для этой области ( USP18 ) и все остальные зонды хромосомы 22 дистальнее трех амплифицированных зондов (рис. 2а).
Рисунок 2 ( a ) Результат MLPA (черные закрашенные кружки) и результат массива SNP (серые закрашенные кружки) области от 22q11.1 до 22q11.23 у индексного пациента. Различные синдромы, включая CES, связанные с этим хромосомным участком, показаны выше. ( b ) Обзор генов в критической области CES и расположения BAC-зондов RP11-155N18 и RP11-958h30, используемых для FISH, вместе с расположением тройной области у индексного пациента и тройной области пациента Мерс и др. Стрелки внутри генов указывают направление транскрипции.
Стрелки внутри генов указывают направление транскрипции.
Изображение в полный размер
После анализа MLPA было проведено стандартное кариотипирование, чтобы выяснить, была ли частичная амплификация 22-й хромосомы вызвана наличием дополнительной маркерной хромосомы. Окрашивание GTG выявило микроскопически нормальный женский кариотип в 10 метафазах.
Метафазный анализ FISH с использованием двух зондов, специфичных для региона CES, показал специфические сигналы только в полосе q11 обеих хромосом 22. В некоторых метафазах RP11-9Сигнал 58h30 казался больше на одной хромосоме 22, чем на другом гомологе, но сигналы не разделялись (рис. 3b). RP11-155N18 не показал четкой разницы в силе сигнала между обоими гомологами.
Рисунок 3 ( a ) Метафазное распространение и ( b ) интерфазные ядра, контрастирующие с DAPI, после гибридизации с RP11-958h30 (зеленый) и p190.22 (красный, специфичный для центромерной области хромосомы 22) ). Зеленые стрелки показывают больший сигнал для RP11-958h30 в метафазных хромосомах и расщепленный сигнал в интерфазных ядрах, при котором сигнал расщепляется на больший и меньший сигнал. ( c ) Схематическое изображение предлагаемого расположения трипликации в хромосоме 22 с красным и зеленым расположением зондов FISH, используемых на панелях ( a , b ).
Зеленые стрелки показывают больший сигнал для RP11-958h30 в метафазных хромосомах и расщепленный сигнал в интерфазных ядрах, при котором сигнал расщепляется на больший и меньший сигнал. ( c ) Схематическое изображение предлагаемого расположения трипликации в хромосоме 22 с красным и зеленым расположением зондов FISH, используемых на панелях ( a , b ).
Полноразмерное изображение
Однако интерфазный анализ FISH показал две копии RP11-155N18 с разной интенсивностью в каждом ядре, тогда как 12 из 30 ядер показали расщепленный сигнал большего из двух сигналов для RP11-958h30 (рис. 3b). ), что приводит к трем сигналам в интерфазной ячейке. В клетках, показывающих три сигнала, наблюдалась дополнительная разница в интенсивности между сигналами. Полученный кариотип был обозначен как 46,XX.ish 22q11.1q11.21(RP11-958х30,РП11-155Н18)х2[10].ядерный 22q11.1(РП11-155Н18х2)[29],22q11.21(РП11-958х30х3)[12/30].
Затем был проведен анализ массива SNP с целевым анализом области 22q11 для определения точного размера амплификации. Массив показал амплификацию 421 зонда в области 598 т.п.н., начиная с 16 179 398 пн в полосе q11.1 и заканчивая 16 777 897 пн в полосе q11.21 (геномная сборка NCBI36/hg18. Геномная сборка GRCh47/hg19 : позиция с 17 799 398 по 18 397 897 п.н.) с оценочным числом копий 4, в соответствии с результатом MLPA (рис. 2а). В амплификации участвовали гены CECR2 , SLC25A18 , ATP6V1E1 , BCL2L13 , BID , все полностью и MICAL3 частично (рис. 2b).
Массив показал амплификацию 421 зонда в области 598 т.п.н., начиная с 16 179 398 пн в полосе q11.1 и заканчивая 16 777 897 пн в полосе q11.21 (геномная сборка NCBI36/hg18. Геномная сборка GRCh47/hg19 : позиция с 17 799 398 по 18 397 897 п.н.) с оценочным числом копий 4, в соответствии с результатом MLPA (рис. 2а). В амплификации участвовали гены CECR2 , SLC25A18 , ATP6V1E1 , BCL2L13 , BID , все полностью и MICAL3 частично (рис. 2b).
Оценка других членов семьи с фенотипическими характеристиками CES с использованием теста 22q11 MLPA показала наследование амплификации у четырех членов семьи и трех поколений (рис. 1) с аналогичным размером и соотношением сигналов.
Обсуждение
Мы описываем четырех членов семьи из трех поколений, у которых с помощью MLPA был обнаружен атипичный прирост части критической области CES. Соотношение сигналов аберрантных зондов указывало на тетрасомию этой части хромосомы 22 (рис. 2а). Стандартное кариотипирование исключало наличие нештатной маркерной хромосомы, обычно обнаруживаемой при СЭС. Метафазный анализ FISH исключил вставку амплифицированного фрагмента в другую хромосому или участок хромосомы. Следовательно, мы пришли к выводу, что имеет место локальная амплификация в одной из хромосом 22. Это подтверждается интерфазными результатами FISH, поскольку больший из двух сигналов для зонда RP11-958 ч 30 мин было разделено на две части в 12 из 30 ядер (рис. 3б).
Метафазный анализ FISH исключил вставку амплифицированного фрагмента в другую хромосому или участок хромосомы. Следовательно, мы пришли к выводу, что имеет место локальная амплификация в одной из хромосом 22. Это подтверждается интерфазными результатами FISH, поскольку больший из двух сигналов для зонда RP11-958 ч 30 мин было разделено на две части в 12 из 30 ядер (рис. 3б).
Используя массив SNP с высоким разрешением, мы обнаружили, что амплификация была ограничена дистальной частью критической области CES и составила около 600 kb (рис. 2a). Четыре сигнала, как и можно было ожидать при интерфазной FISH на основании результатов MLPA и массива, не наблюдались, вероятно, из-за близкого физического расположения различных копий на аберрантной хромосоме 22. Примерно в половине исследованных ядер три сигнала для Датчик BAC RP11-958:30 наблюдались. Наблюдалась четкая разница в интенсивности трех сигналов, как показано на рисунке 3b. BAC-зонд RP11-958h30 расположен на проксимальном конце амплифицированного фрагмента размером 600 т. п.н. Следовательно, мы заключаем, что не только три копии фрагмента присутствуют в одном и том же локусе аберрантной хромосомы 22, но также и то, что средний фрагмент имеет инвертированную ориентацию по сравнению с двумя фланкирующими, как предполагается на рис. 3с. Следовательно, это приведет к одному раздельному сигналу и одному комбинированному, двойному сигналу. Тот факт, что три копии фрагмента размером 600 т.п.н. присутствуют на одной и той же хромосоме, также подтверждается (аутосомно-доминантным) типом наследования в семье (рис. 1).
п.н. Следовательно, мы заключаем, что не только три копии фрагмента присутствуют в одном и том же локусе аберрантной хромосомы 22, но также и то, что средний фрагмент имеет инвертированную ориентацию по сравнению с двумя фланкирующими, как предполагается на рис. 3с. Следовательно, это приведет к одному раздельному сигналу и одному комбинированному, двойному сигналу. Тот факт, что три копии фрагмента размером 600 т.п.н. присутствуют на одной и той же хромосоме, также подтверждается (аутосомно-доминантным) типом наследования в семье (рис. 1).
Принятая в настоящее время критическая область CES основана на одном пациенте со всеми кардинальными признаками CES, вызванными атипичной добавочной кольцевой хромосомой 22. 13 Эта атипичная добавочная маркерная хромосома не включала амплификацию непосредственно дистально расположенных генов BCL2L13 , BID и MICAL3 (рис. 2b). Однако эти гены амплифицируются в более часто встречающейся сверхштатной маркерной хромосоме стандартного типа у пациентов с CES. Как у первоначального пациента, описанного Мирсом et al 13 умерли на 17-й день жизни, нельзя исключать вклад в физическое и умственное развитие этих дистально расположенных генов BCL2L13 , BID и MICAL3 . 12
Как у первоначального пациента, описанного Мирсом et al 13 умерли на 17-й день жизни, нельзя исключать вклад в физическое и умственное развитие этих дистально расположенных генов BCL2L13 , BID и MICAL3 . 12
В амплификации, обнаруженном в нынешнем семействе, шесть генов, CECR2 , SLC25A18 , ATP6V1E1 , BCL2L13 , BID и MCAL3 . Члены семьи, несущие внутрихромосомную амплификацию, имеют анальную атрезию (3 из 4) и/или преаурикулярные ямки или метки (4 из 4). Поскольку эти врожденные аномалии часто обнаруживаются при CES и поскольку только три гена, участвующие в нашей семье, перекрываются с генами, участвующими в амплификации, о которой сообщает Mears et al. 13 ( CECR2 , SLC25A18 и ATP6V1E1 ) (рис. 2b), атрезия анального отверстия и преаурикулярные ямки или метки могут быть вызваны амплификации одного или нескольких из этих генов и/или сверхэкспрессией. Те же гены могут быть также ответственны за дефицит гормона роста и одностороннюю агенезию почки, как это наблюдалось у дяди пациента-индекса, потому что низкий рост и аномалии почек также являются частью фенотипического спектра CES. Ранее сообщалось о пациенте с CES с низким уровнем гормона роста в сыворотке и гипогонадотропным гипогонадизмом. 16 Кроме того, мы не можем исключить, что даже глухота и врожденные аномалии лица у двоюродного дедушки основного пациента, ранее клинически диагностированного как синдром Гольденхара, вызваны той же семейной тройственностью. К сожалению, дедушка пациента-индекса отказался от дальнейшего тестирования на возможное носительство. Эти гены также могут быть вовлечены в другие фенотипические особенности CES, которые могут быть непенетрантными в этом семействе.
Те же гены могут быть также ответственны за дефицит гормона роста и одностороннюю агенезию почки, как это наблюдалось у дяди пациента-индекса, потому что низкий рост и аномалии почек также являются частью фенотипического спектра CES. Ранее сообщалось о пациенте с CES с низким уровнем гормона роста в сыворотке и гипогонадотропным гипогонадизмом. 16 Кроме того, мы не можем исключить, что даже глухота и врожденные аномалии лица у двоюродного дедушки основного пациента, ранее клинически диагностированного как синдром Гольденхара, вызваны той же семейной тройственностью. К сожалению, дедушка пациента-индекса отказался от дальнейшего тестирования на возможное носительство. Эти гены также могут быть вовлечены в другие фенотипические особенности CES, которые могут быть непенетрантными в этом семействе.
Мутации в гене CECR2 вызывает дисрегуляцию мезенхимальных или эктодермальных факторов транскрипции, и описано, что этот белок способствует нейрогенезу и развитию внутреннего уха. 17, 18 SLC25A18 катализирует однонаправленный транспорт глутамата во внутренних мембранах митохондрий. 19 ATP6V1E1 кодирует вспомогательную регуляторную субъединицу E Н(+)-АТФазы вакуолярного типа, которая отвечает за подкисление внутриклеточных органелл и участвует в транспорте ионов водорода через плазматическую мембрану. Было показано, что мутации в других субъединицах комплекса V-АТФазы участвуют в ацидозе дистальных почечных канальцев и сенсоневральной глухоте. 20
17, 18 SLC25A18 катализирует однонаправленный транспорт глутамата во внутренних мембранах митохондрий. 19 ATP6V1E1 кодирует вспомогательную регуляторную субъединицу E Н(+)-АТФазы вакуолярного типа, которая отвечает за подкисление внутриклеточных органелл и участвует в транспорте ионов водорода через плазматическую мембрану. Было показано, что мутации в других субъединицах комплекса V-АТФазы участвуют в ацидозе дистальных почечных канальцев и сенсоневральной глухоте. 20
В заключение, CECR2 , SLC25A18 и ATP6V1E1 являются сильными генами-кандидатами, вызывающими анальную атрезию, преаурикулярные ямки или метки и почечные аномалии у описанных членов семейства CES и, следовательно, вызывающие эти частые компоненты CES. спектр фенотипа.
Ссылки
Belangero SI, Bellucco FT, Cernach MC, Hacker AM, Emanuel BS, Melaragno MI: Прерванная дуга аорты типа B у пациента с синдромом кошачьего глаза.
: e29–e31, e56-28. Бюстгальтеры Arq Cardiol 2009; 92
Бюстгальтеры Arq Cardiol 2009; 92 Артикул Google ученый
Берендс М.Дж., Тан-Синдхуната Г., Легте Б., ван Эссен А.Дж.: Фенотипическая изменчивость синдрома кошачьего глаза. Genet Couns 2001; 12 : 23–34.
КАС пабмед Google ученый
Rosias PR, Sijstermans JM, Theunissen PM и др. : Фенотипическая изменчивость синдрома кошачьего глаза. Клинический случай и обзор литературы. Genet Couns 2001; 12 : 273–282.
КАС пабмед Google ученый
Buhler EM, Mehes K, Muller H, Stalder GR : Синдром кошачьего глаза, частичная трисомия 22. Humangenetik 1972; 15 : 150–162.
КАС пабмед Google ученый
Schinzel A, Schmid W, Fraccaro M
Артикул КАС Google ученый
Reiss JA, Weleber RG, Brown MG, Bangs CD, Lovrien EW, Magenis RE: Тандемная дупликация проксимального отдела 22q: причина синдрома кошачьего глаза. Am J Med Genet 1985; 20 : 165–171.
Артикул КАС Google ученый
Нолл Дж. Х., Асамоа А., Плетчер Б. А., Вагстафф Дж. Интерстициальная дупликация проксимального участка 22q: фенотипическое совпадение с синдромом кошачьего глаза. Am J Med Genet 1995; 55 : 221–224.
Артикул КАС Google ученый
Мейнс М., Бурфайнд П., Мотш С и др. : Частичная трисомия хромосомы 22 в результате интерстициальной дупликации 22q11.2 у ребенка с типичным синдромом кошачьего глаза.
J Med Genet 2003; 40 : е62.Артикул КАС Google ученый
Lindsay EA, Shaffer LG, Carrozzo R, Greenberg F, Baldini A: De novo тандемная дупликация сегмента хромосомы 22q11-q12: клиническая, цитогенетическая и молекулярная характеристика. Am J Med Genet 1995; 56 : 296–299.
Артикул КАС Google ученый
Артикул КАС Google ученый
Роза Р.Ф., Момбах Р., Зен П.Р., Грациадио С., Паскулин Г.А. Клинические характеристики выборки пациентов с синдромом кошачьего глаза. Rev Assoc Med Bras 2010; 56 : 462–465.
Артикул Google ученый
Footz TK, Brinkman-Mills P, Banting GS et al : Анализ критической области синдрома кошачьего глаза у людей и области консервативной синтении у мышей: поиск генов-кандидатов на хромосоме человека или рядом с ней 22 перицентромер. Геном Res 2001; 11 : 1053–1070.
Артикул КАС Google ученый
Мирс А.Дж., Эль-Шанти Х., Мюррей Дж.С., Макдермид Х.Э., Патил С.Р.: Незначительная дополнительная кольцевая хромосома 22, связанная с синдромом кошачьего глаза: дальнейшее определение критической области. Am J Hum Genet 1995; 57 : 667–673.
КАС пабмед ПабМед Центральный Google ученый
van der Veken LT, Dieleman MM, Douben H et al : Мозаика низкой степени для сложной нештатной кольцевой хромосомы 18 у взрослого пациента с множественными врожденными аномалиями.
Мол Цитогенет 2010; 3 : 13.Артикул Google ученый
Рокки М., Архидиаконо Н., Антоначчи Р. и др. : Клонирование и сравнительное картирование недавно созданной человеческой хромосомы 22-специфичной альфа-сателлитной ДНК. Somat Cell Mol Genet 1994; 20 : 443–448.
Артикул КАС Google ученый
Масукава Х., Одзаки Т., Ногимори Т. Синдром кошачьего глаза с гипогонадотропным гипогонадизмом. Internal Med 1998; 37 : 853–856.
Артикул КАС Google ученый
Fairbridge NA, Dawe CE, Niri FH, Kooistra MK, King-Jones K, McDermid HE : Мутации Cecr2, вызывающие экзэнцефалию, вызывают нарушение регуляции мезенхимальных/эктодермальных факторов транскрипции. Врожденные дефекты Res A Clin Mol Teratol 2010; 88 : 619–625.
Артикул КАС Google ученый
Dawe CE, Kooistra MK, Fairbridge NA, Pisio AC, McDermid HE: Роль гена ремоделирования хроматина Cecr2 в нейруляции и развитии внутреннего уха. Дев Дин 2011; 240 : 372–383.
Артикул КАС Google ученый
Fiermonte G, Palmieri L, Todisco S, Agrimi G, Palmieri F, Walker JE: Идентификация митохондриального переносчика глутамата. Бактериальная экспрессия, восстановление, функциональная характеристика и распределение в тканях двух изоформ человека. J Biol Chem 2002; 277 : 19289–19294.
Артикул КАС Google ученый
Stover EH, Borthwick KJ, Bavalia C et al : Новые мутации ATP6V1B1 и ATP6V0A4 при аутосомно-рецессивном ацидозе дистальных почечных канальцев с новыми доказательствами потери слуха.
J Med Genet 2002; 39 : 796–803.Артикул КАС Google ученый
 Бюстгальтеры Arq Cardiol 2009; 92
Бюстгальтеры Arq Cardiol 2009; 92  Отчет 11 пациентов и описание клинической картины. Hum Genet 1981; 57 : 148–158.
Отчет 11 пациентов и описание клинической картины. Hum Genet 1981; 57 : 148–158. J Med Genet 2003; 40 : е62.
J Med Genet 2003; 40 : е62.
 Мол Цитогенет 2010; 3 : 13.
Мол Цитогенет 2010; 3 : 13.
 J Med Genet 2002; 39 : 796–803.
J Med Genet 2002; 39 : 796–803.СПРАВЕДЕНИЯ СПИСОК ЛИТЕРАТУРЫ
Информация о авторе
Авторы и принадлежности
Департамент клинической генетики, Erasmus Medical Center, Rotterdam, Netherlands
Jeroen Knijnergan, vorelterte -vanemal, varentemal, varentemal, varentem. varentem. Galhana M Bolman, Rosa Laura E van Loon, H Berna Beverloo и Laura JCM van Zutven
Авторы
- Jeroen Knijnenburg
Посмотреть публикации авторов
Вы также можете искать этого автора в PubMed Google Scholar
- Yolande van Bever
Посмотреть публикации автора
Вы также можете искать этого автора в PubMed Google Scholar
- Lorette O M Hulsman
Просмотр публикаций автора
Вы также можете искать этого автора в PubMed Google Scholar
- Chantal AP van Kempen
Посмотреть публикации автора
Вы также можете искать этого автора в PubMed Google Scholar
- Galhana M Bolman
Посмотреть публикации автора
Вы также можете искать этого автора в PubMed Google Scholar
- Rosa Laura E van Loon
Посмотреть публикации автора
Вы также можете искать этого автора в PubMed Google Scholar
- H Berna Beverloo
Посмотреть публикации автора
Вы также можете искать этого автора в PubMed Google Scholar
- Laura JCM van Zutven
Просмотр публикаций автора
Вы также можете искать этого автора в PubMed Google Scholar
Автор, ответственный за переписку
Лаура Дж. К. М. ван Зутвен.
К. М. ван Зутвен.
Заявление об этике
Конкурирующие интересы
Авторы заявляют об отсутствии конфликта интересов.
Права и разрешения
Перепечатки и разрешения
Об этой статье
Синдром Фрейзера — NORD (Национальная организация по редким заболеваниям) Joel Noutakdie Tochie, доктору медицинских наук, отделение анестезиологии и реанимации, факультет медицины и биомедицинских наук, Университет Яунде I, Камерун, за помощь в подготовке этого отчета.
Synonyms of Fraser Syndrome
- cryptophthalmos-syndactyly syndrome
- cryptophthalmos syndrome
- cryptophthalmos with other malformations
- Fraser-Francois syndrome
- Meyer-Schwickerath syndrome
- Ulrich-Feichtiger syndrome
- FS
Subdivisions of Fraser Синдром
- Синдром Фрейзера 1 (FRASRS1)
- Синдром Фрейзера 2 (FRASRS2)
- Синдром Фрейзера 3 (FRASRS3)
Признаки и симптомы
Причины
Синдром Фрейзера вызывается изменениями (мутациями) в генах FRAS1 , FREM1 , FREM2 или GRIP6 90). Более конкретно, синдром Фрейзера 1 (FRASRS1) вызывается мутациями гена субъединицы 1 комплекса внеклеточного матрикса Фрейзера ( FRAS1 ). Синдром Фрейзера 2 (FRASRS2) вызывается мутациями в гене 2 белка внеклеточного матрикса, связанного с FRAS1 ( FREM2 ). Синдром Фрейзера 3 (FRASRS3) вызывается мутациями белка 1, взаимодействующего с рецептором глутамата (9).0555 GRIP1 ) ген. Мутации гена FRAS1 являются наиболее частой причиной синдрома Фрейзера, что составляет примерно половину случаев FS; тогда как мутации гена FREM2 и мутации гена GRIP1 составляют меньший процент случаев.
Более конкретно, синдром Фрейзера 1 (FRASRS1) вызывается мутациями гена субъединицы 1 комплекса внеклеточного матрикса Фрейзера ( FRAS1 ). Синдром Фрейзера 2 (FRASRS2) вызывается мутациями в гене 2 белка внеклеточного матрикса, связанного с FRAS1 ( FREM2 ). Синдром Фрейзера 3 (FRASRS3) вызывается мутациями белка 1, взаимодействующего с рецептором глутамата (9).0555 GRIP1 ) ген. Мутации гена FRAS1 являются наиболее частой причиной синдрома Фрейзера, что составляет примерно половину случаев FS; тогда как мутации гена FREM2 и мутации гена GRIP1 составляют меньший процент случаев.
Гены содержат инструкции по созданию белков, играющих важную роль во многих функциях организма. Когда происходит мутация гена, белковый продукт может быть дефектным, неэффективным, отсутствовать или вырабатываться в избытке. В зависимости от функций конкретного белка это может повлиять на многие системы органов тела, включая мозг.
Гены FRAS1 и FREM2 продуцируют белки (белки FRAS1 и FREM2 соответственно), которые работают вместе как часть комплекса FRAS/FREM. Одна из функций комплекса FRAS/FREM состоит в том, чтобы соединить различные слои кожи вместе, один поверх другого, чтобы, по сути, сформировать кожу. Его функция важна в эмбриональный период до рождения. Эта группа белков играет роль в правильном развитии кожи, внутренних органов, включая почки и другие ткани. Таким образом, мутация в 9Ген 0555 FRAS1 или FREM2 вызывает неисправность соответствующего белка, поэтому комплекс FRAS/FREM не может функционировать должным образом и, следовательно, приводит к неправильному развитию кожи, внутренних органов и других тканей. Это неправильное развитие в конечном итоге вызывает такие признаки и симптомы, как криптофтальм, кожная синдактилия и агенезия почек. Ген GRIP1 продуцирует белок GRIP1, который гарантирует, что белки FRAS1 и FREM2 попадают в правильное место внутри клетки для выполнения своей функции. Мутации в Ген GRIP1 препятствует нормальному функционированию белков FRAS1 и FREM2, вызывая неправильное развитие кожи, внутренних органов и других тканей.
Одна из функций комплекса FRAS/FREM состоит в том, чтобы соединить различные слои кожи вместе, один поверх другого, чтобы, по сути, сформировать кожу. Его функция важна в эмбриональный период до рождения. Эта группа белков играет роль в правильном развитии кожи, внутренних органов, включая почки и другие ткани. Таким образом, мутация в 9Ген 0555 FRAS1 или FREM2 вызывает неисправность соответствующего белка, поэтому комплекс FRAS/FREM не может функционировать должным образом и, следовательно, приводит к неправильному развитию кожи, внутренних органов и других тканей. Это неправильное развитие в конечном итоге вызывает такие признаки и симптомы, как криптофтальм, кожная синдактилия и агенезия почек. Ген GRIP1 продуцирует белок GRIP1, который гарантирует, что белки FRAS1 и FREM2 попадают в правильное место внутри клетки для выполнения своей функции. Мутации в Ген GRIP1 препятствует нормальному функционированию белков FRAS1 и FREM2, вызывая неправильное развитие кожи, внутренних органов и других тканей.
Синдром Фрейзера наследуется по аутосомно-рецессивному типу. Рецессивные генетические нарушения возникают, когда человек наследует один и тот же аномальный/мутированный ген одного и того же признака от каждого родителя. Если человек получает один нормальный ген и один ген болезни, он будет носителем болезни, но обычно не будет проявлять симптомов. Риск для двух родителей-носителей передать дефектный ген и, следовательно, родить больного ребенка составляет 25% при каждой беременности. Риск рождения ребенка-носителя, как и родители, составляет 50% при каждой беременности. Вероятность того, что ребенок получит нормальные гены от обоих родителей и будет генетически нормальным по этому конкретному признаку, составляет 25%. Риск одинаков для мужчин и женщин.
Все люди несут несколько аномальных генов. У родителей, которые являются близкими родственниками (родственными родственниками), больше шансов, чем у неродственных родителей, нести один и тот же аномальный ген, что увеличивает риск рождения детей с рецессивным генетическим заболеванием.
Пораженные группы населения
Синдром Фрейзера поражает мужчин и женщин в равной степени. Возраст начала этого расстройства — неонатальный / антенатальный, что означает, что у пострадавшего человека это расстройство развивается до рождения. Считается, что FS более распространен среди цыганского населения (этническая принадлежность рома) в южной и восточной Европе.
Частота ФС составляет 0,43 на 10 000 или 1 на 200 000 новорожденных и 11,06 на 10 000 новорожденных, умерших до рождения (мертворождения).
Диагностика
Синдром Фрейзера можно диагностировать до рождения с помощью УЗИ на 18-й неделе беременности. Пренатальная ультразвуковая диагностика обычно проводится при наличии семейного анамнеза заболевания. Диагноз можно поставить, если на УЗИ плода присутствуют два из следующих признаков: микрофтальмия (аномально малый размер одного глаза), синдактилия, увеличение эхогенности легких, маловодие (дефицит околоплодных вод во время беременности).
Синдром Фрейзера обычно диагностируется при рождении на основании признаков и симптомов, обнаруженных у ребенка. IT Thomas и его коллеги предложили следующие критерии для диагностики ФС: наличие одного большого критерия и одного малого критерия или, альтернативно, наличие двух больших критериев и одного малого критерия. Основные критерии включают кожную синдактилию, криптофтальм, аномалии наружных половых органов, аноректальные аномалии и аномалии конечностей. Малыми критериями являются аномалии уха и носа, дефекты костей черепа, пупочная грыжа, умственная отсталость и аномалии мочевыводящих и дыхательных путей. Ни один из основных критериев не является обязательным для постановки диагноза.
Генетическое тестирование на наличие мутаций в генах FRAS1 , FREM1 , FREM2 или GRIP1 может подтвердить диагноз синдрома Фрейзера.
Стандартная терапия
Лечение
В настоящее время нет лекарства от синдрома Фрейзера. Лечение FS может включать хирургическое вмешательство для исправления некоторых пороков развития, связанных с этим заболеванием, в зависимости от их тяжести. Другое лечение является симптоматическим и поддерживающим. Для оценки каждого пациента и определения способов лечения симптомов требуется группа специалистов. Прогноз без лечения зависит от тяжести конкретных аномалий у пациента, особенно пороков развития дыхательных путей и анальных имперфораций. К сожалению, у детей с тяжелыми аномалиями часто встречается смерть на первом году жизни.
Лечение FS может включать хирургическое вмешательство для исправления некоторых пороков развития, связанных с этим заболеванием, в зависимости от их тяжести. Другое лечение является симптоматическим и поддерживающим. Для оценки каждого пациента и определения способов лечения симптомов требуется группа специалистов. Прогноз без лечения зависит от тяжести конкретных аномалий у пациента, особенно пороков развития дыхательных путей и анальных имперфораций. К сожалению, у детей с тяжелыми аномалиями часто встречается смерть на первом году жизни.
Семьям детей с этим заболеванием рекомендуется генетическое консультирование.
Investigational Therapies
Информация о текущих клинических испытаниях размещена в Интернете на сайте www.clinicaltrials.gov. Все исследования, финансируемые правительством США, а некоторые из них поддерживаются частным сектором, публикуются на этом правительственном веб-сайте.
Для получения информации о клинических испытаниях, проводимых в Клиническом центре NIH в Бетесде, штат Мэриленд, обращайтесь в отдел набора пациентов NIH:
Бесплатный звонок: (800) 411-1222
Телетайп: (866) 411-1010
Электронная почта: [email protected]
Некоторые текущие клинические испытания также размещены на следующей странице веб-сайта NORD: https://rarediseases. org/for-patients-and-families/information-resources/info-clinical-trials-and-research-studies/
org/for-patients-and-families/information-resources/info-clinical-trials-and-research-studies/
Для получения информации о клинических испытаниях, спонсируемых частными источниками, обращайтесь: http://www.centerwatch.com/
Для получения информации о клинических испытаниях, проведенных в Европе, обращайтесь: https://www.clinicaltrialsregister.eu/
Ссылки
УЧЕБНИКИ
Stevens, C. In NORD Guide to Rare Disorders. Липпинкотт Уильямс и Уилкинс. Филадельфия, Пенсильвания. 2003:196.
СТАТЬИ В ЖУРНАЛЕ
Alsaman MZB, Agha S, Sallah H, Badawi R, Kitaz MN, Assani A, Nawfal H. Двусторонняя анофтальмия и внутрипеченочная билиарная атрезия, два необычных компонента синдрома Фрейзера: клинический случай. BMC Беременность и роды 2020; 20: 358.
Мбонда А., Эндомба Ф.Т., Канмунье США, Нкек Дж.Р., Точи Дж.Н. Диагноз синдрома Фрейзера был пропущен до шестимесячного возраста в условиях ограниченных ресурсов: история болезни. БМЦ Педиатрия. 2019;19(1):292.
Кришнан Д., Авабрата К., D«SQ» Соуза А. Синдром Фрейзера. Журнал медицинских наук и исследований Мюллера. 2014;5(1):85-85.
Баришич И., Одак Л., Лоан М. и др. Синдром Фрейзера: эпидемиологическое исследование европейского населения. Американский журнал медицинской генетики, часть A. 2013; 161 (5): 1012-1018.
Каланити К., Сандья В. Синдром Фрейзера у трех последовательных братьев и сестер. Оман Дж. Офтальмол. 2011;4(2):87-89.
Окумус Н., Онал Э.Е., Туркилмаз С. и др. Неэффективность реанимационных мероприятий у новорожденного, недиагностированного во внутриутробном периоде. Реанимация. 2005;65:221-23.
Вонг Л.Дж., Лин Ю.Х. Суваннарат П. и др. Мутации митохондриальной ДНК у пациента с изменением пола и клиническими признаками, соответствующими синдрому Фрейзера. Клин Жене. 005;67:252-57.
Хамбире С.Д., Бхавсар П.П., Джаякар А.В. Синдром Фрейзера-криптофтальма с сердечно-сосудистыми пороками развития: редкая ассоциация. Индийский педиатр. 2003;40:888-90.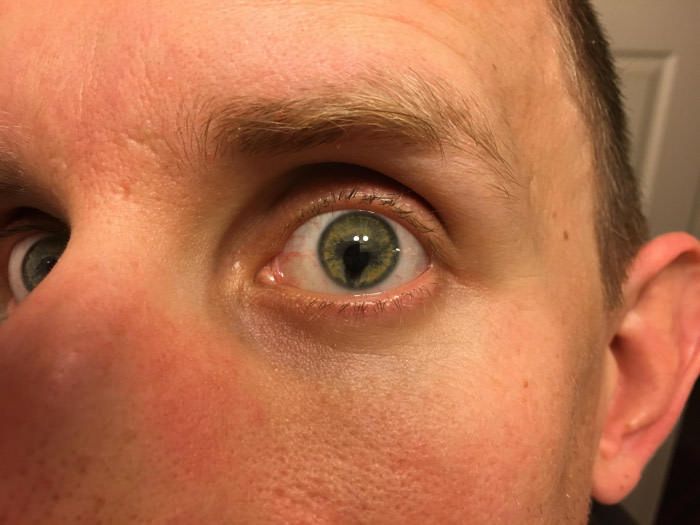
Вронтоу С., Петроу П., Мейер Б.И. и др. Дефицит Fras1 приводит к криптофтальму, агенезии почек и пузырчатому фенотипу у мышей. Нат Жене. 2003;34:209-14.
МакГрегор Л., Макела В., Дарлинг С.М. и др. Синдром Фрейзера и фенотип мышиных пузырей, вызванные мутациями в FRAS1/Fras1, кодирующем предполагаемый белок внеклеточного матрикса. Нат Жене. 2003;34:203-08.
Руссо Т., Лоран Н., Товен-Робинет С. и др. Пренатальная диагностика и внутрисемейная клиническая гетерогенность синдрома Фрейзера. Пренат Диагн. 2002; 22:692-96.
Славотинек А.М., Тиффт С.Дж. Синдром Фрейзера и криптофтальм: обзор диагностических критериев и данных о фенотипических модулях при сложных синдромах пороков развития. J Med Genet. 2002;39: 623-33.
Томас И.Т., Фриас Дж.Л., Феликс В., Санчес де Леон Л., Эрнандес Р.А., Джонс М.С. Изолированный и синдромальный криптофтальм. Am J Med Genet. 1986; 25(1):85–98.
ИНТЕРНЕТ
Синдром Фрейзера – Домашний справочник по генетике – NIH. Национальная медицинская библиотека. Опубликовано 17 августа 2020 г. https://ghr.nlm.nih.gov/condition/fraser-syndrome#. По состоянию на 30 августа 2020 г.
Национальная медицинская библиотека. Опубликовано 17 августа 2020 г. https://ghr.nlm.nih.gov/condition/fraser-syndrome#. По состоянию на 30 августа 2020 г.
Martinez-Frias ML, Bermizo Sanchez E, Felix V. Частота синдрома Фрейзера в нашей среде 189 и клинические эпидемиологические аспекты последовательных серий случаев. ЭСП 190 Педиатр 1998; 48: 634-38.
Неоднозначные гениталии: Медицинская энциклопедия MedlinePlus. Обновлено 4 августа 2020 г. https://medlineplus.gov/ency/article/003269.htm. По состоянию на 30 августа 2020 г.
СИНДРОМ ФРЕЙЗЕРА 1; FRASRS1. OMIM.Обновлено 26 декабря 2019 г. https://omim.org/entry/219000#description По состоянию на 30 августа 2020 г.
Синдром Фрейзера. ГолубьМед. 18 мая 2014 г. Обновлено 15 сентября 2018 г. https://www.dovemed.com/diseases-conditions/fraser-syndrome/. По состоянию на 30 августа 2020 г.
Синдром Фрейзера. Информационный центр генетических и редких заболеваний. Обновлено 7 декабря 2016 г. https://rarediseases. info.nih.gov/diseases/6465/fraser-syndrome#ref_12722. По состоянию на 30 августа 2020 г.
info.nih.gov/diseases/6465/fraser-syndrome#ref_12722. По состоянию на 30 августа 2020 г.
Синдром Фрейзера. сирота. Обновлено в марте 2006 г. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=2052. По состоянию на 30 августа 2020 г.
Заячья губа и волчья пасть – симптомы и причины. Клиника Майо. Опубликовано 22 мая 2018 г. https://www.mayoclinic.org/diseases-conditions/cleft-palate/symptoms-causes/syc-20370985 По состоянию на 30 августа 2020 г.
Онтология фенотипов человека. https://hpo.jax.org/app/browse/disease/OMIM:219000. По состоянию на 30 августа 2020 г.
Годы публикации
1989, 1997, 2006, 2020
Информация в базе данных NORD по редким заболеваниям предназначена только для образовательных целей и не предназначена для замены рекомендаций врача или другого квалифицированного медицинского работника.
Содержание веб-сайта и баз данных Национальной организации редких заболеваний (NORD) защищено авторским правом и не может быть воспроизведено, скопировано, загружено или распространено каким-либо образом в коммерческих или общественных целях без предварительного письменного разрешения и одобрения.