Прогрессирующая деменция: 404 | Elu Dementsusega
Прогрессирующая деменция | Пансионат для пожилых людей
Для пожилого возраста характерно развитие патологий неврологического характера. Наиболее серьезной и часто встречающейся является деменция, или старческое слабоумие.
Если верить статистике, этому недугу подвержено свыше 70 % людей преклонного возраста. С каждым годом эта цифра растет. Данный патологический процесс проявляется по-разному, то есть имеет различные степени тяжести. Но данный диагноз чаще всего называют не иначе как прогрессирующей деменцией. Дело в том, что патология все время развивается, что ухудшает состояние пациента и качество его жизни.
Содержание:
- Как проявляет себя деменция
- Что представляет собой болезнь
- Из-за чего развивается деменция
- Способы борьбы с деменцией
Как проявляет себя деменция
Пожилые люди, страдающие прогрессирующей деменцией, сталкиваются с множеством тяжелых нарушений, которые оказывают влияние на их психологическое и физическое состояние. Современные возможности позволяют приостановить развитие патологического процесса. Проще всего это сделать на ранних стадиях, когда нарушения еще не носят фатальный характер.
Важно: своевременное обращение к врачу поможет вовремя выявить деменцию и остановить ее прогрессирование. Для проведения грамотного лечения необходимо точно поставить диагноз. Правильно проанализировать все климатические признаки и собрать анамнез может только специалист.
Главными симптомами старческой деменции являются:
- ухудшение памяти. Этот признак считают одним из первых проявлений недуга. На начальных стадиях симптом имеет слабовыраженный характер, но со временем способен стремительно прогрессировать. На последних стадиях деменции пациенты часто забывают имена и лица членов семьи, важные события и даты;
- нарушение речи. На начальном этапе развития заболевания человек испытывает сложности с тем, чтобы подобрать подходящее слово или правильно назвать предмет.
 Со временем больной растягивает фразы или бубнит, теряет способность выразить мысли словами;
Со временем больной растягивает фразы или бубнит, теряет способность выразить мысли словами; - снижение концентрации внимания. Больной не может сосредоточиться на мысли или действии. Постепенно развивается заторможенность — чаще всего она выражается тем, что больной смотрит в одну точку, однако мыслительные процессы не происходят;
- психические расстройства. По мере развития деменции пациент становится неуравновешенным. На первых стадиях близкие отмечают резкие смены настроения. Человек становится более злым, замкнутым и ворчливым. Со временем это состояние усугубляется: люди, страдающие данным недугом, часто находятся в депрессивном состоянии, они подвержены апатии, бредят;
- тремор. Дрожание ног и рук говорит о том, что деменция стремительно прогрессирует.
 Со временем больной растягивает фразы или бубнит, теряет способность выразить мысли словами;
Со временем больной растягивает фразы или бубнит, теряет способность выразить мысли словами;Что представляет собой болезнь
Для остановки прогрессирования деменции в первую очередь нужно знать о природе данного заболевания и механизмах его развития.
Деменцией в медицине называют угасание высшей нервной деятельности. Происхождение патологического процесса является органическим — иными словами, причиной нарушения становится то, что клетки головного мозга теряют способность к регенерации. Кроме того, к развитию деменции приводит разрушение нейронных связей и отдельных нейронов.
Вместе с тем прогрессирующую деменцию также часто считают психическим расстройством. В данном случае степень тяжести клинических признаков напрямую зависит от того, насколько масштабны органические нарушения головного мозга. В процессе развития недуга затрагиваются такие когнитивные функции, как память, речь, а также другие интеллектуальные способности. Прогрессирование патологии приводит к тому, что в итоге происходит полное разрушение личности.
Поставить точный диагноз могут специалисты пансионата «Доверие». Запишите пожилого родственника к нашим врачам на обследование.
Из-за чего развивается деменция
Угасание регенерационных способностей — естественное явление, знаменующее собой приход старости. Этот процесс способствует развитию патологий в полушариях мозга. Этим специалисты и объясняют тот факт, что прогрессирующая деменция бывает у людей преклонного возраста.
Этот процесс способствует развитию патологий в полушариях мозга. Этим специалисты и объясняют тот факт, что прогрессирующая деменция бывает у людей преклонного возраста.
Кроме естественных изменений, есть ряд факторов, которые приводят к развитию деменции у пожилых:
- патологии сердечно-сосудистой системы. К ним относятся различные заболевания сердца и сосудов, однако чаще всего у пациентов с деменцией отмечаются перепады артериального давления;
- травмы позвоночника или головы. Особенно это касается тяжелых повреждений. При этом учитываются травмы, не только полученные в ближайшее время, но и приобретенные в течение всей жизни;
- генетическая предрасположенность. Медики часто называют наследственную предрасположенность одним из основных факторов развития слабоумия. Если у больного в роду были случаи тяжелых психических расстройств или патологий неврологического характера, прогрессирующая деменция даст о себе знать с высокой долей вероятности;
- наличие вредных привычек. В первую очередь речь идет об употреблении спиртного (в особенности при тяжелых формах алкоголизма), однако некоторые специалисты считают, что курение тоже способно привести к этим процессам;
- инфекции. Многие инфекционные заболевания поражают ЦНС и отделы мозга. Самыми опасными являются менингит, энцефалит и нейросифилис;
- инсульты и микроинсульты. У пожилых людей, перенесших подобное явление, часто отмечается прогрессирование деменции, так как заболевание имеет органическое происхождение.
На самом деле факторов, которые приводят к развитию деменции, намного больше — выше мы рассказали о самых распространенных. Следует помнить, что в большинстве случаев знание причины появления патологии помогает назначить эффективное лечение.
Способы борьбы с деменцией
Чтобы победить этот недуг, нужно как можно быстрее, сразу после обнаружения первых признаков и подозрений, обратиться к врачу. Специалист соберет анамнез, проведет диагностику и оценит клинические признаки. Эти действия позволят выявить недуг и определить стадию его развития.
Специалист соберет анамнез, проведет диагностику и оценит клинические признаки. Эти действия позволят выявить недуг и определить стадию его развития.
Важно: в преклонном возрасте невозможно полностью избавиться от прогрессирующей деменции, так как заболевание является неизлечимым. Но грамотная медикаментозная терапия помогает остановить развитие патологии. При своевременном обращении к врачу есть шанс восстановить утраченные навыки и функции.
Так как в процессе развития деменции отмечается усугубление симптомов, многие пациенты со временем теряют навыки самообслуживания, поэтому им необходимы присмотр и тщательный уход. Поскольку в домашних условиях часто сложно организовать подходящую для больного обстановку, стоит поместить его в пансионат.
В частном доме престарелых «Доверие» вашего родного человека обеспечат всем необходимым. Наши специалисты имеют опыт по уходу за больными с деменцией и другими старческими патологиями. Внимательный и вежливый персонал окружит пациента заботой, а врачи проконтролируют состояние здоровья подопечных во время ежедневных обходов. Запишите пожилого родственника к нам на обследование, и мы вместе начнем борьбу с деменцией!
Что такое деменция (слабоумие)? | Билобил
Деменция представляет собой хроническое прогрессирующее заболевание головного мозга. Она характеризуется снижением всех высших когнитивных функций, таких как память, мышление, ориентация, понимание, вычисление, способность учиться, суждение. Развитие деменции сопровождается эмоциональными изменениями: беспокойство, раздражительность, вспыльчивость, снижение самоуважения, депрессия, эмоциональная лабильность, эмоциональная несдержанность, безразличие.
Деменция является общим термином, обозначающим глубокие нарушения памяти и других интеллектуальных способностей. Это серьезное заболевание, которое ставит под угрозу качество повседневной жизни. Есть несколько разных форм деменции; некоторые встречаются чаще, чем другие, хотя все они означают болезнь, которая делает повседневную жизнь гораздо сложнее. К тому же, это заболевание прогрессирует с возрастом.
К тому же, это заболевание прогрессирует с возрастом.
Какая разница между забывчивостью и деменцией?
Как уже упоминалось ранее, забывчивость может наблюдаться у каждого в повседневной жизни. У пожилых людей возрастные проблемы с памятью встречаются гораздо чаще, но не всегда они свидетельствуют о развитии деменции. Для деменции характерно:
- Деменция является общей деградации умственных способностей.
- Пациенты с деменцией не просто забывают зонтик в поезде; они забывают сесть на поезд.
- Они забывают имена своих любимых и путают людей.
- На более поздних стадиях они не узнают даже своих близких или считают совершенно незнакомых людей особами, которых они знали в прошлом.
- Поскольку пациенты совершенно не помнят текущие события, они любят говорить о прошлых событиях, которые они помнят хорошо, по крайней мере, на начальных стадиях деменции.
- Проблемы становятся настолько выраженными, что они ставят под угрозу повседневную жизнь пациентов, лишают их возможности самостоятельно заботиться о себе.
- Пациенты становятся все более и более зависимыми от своих опекунов.
Типичные ответы пациента с деменцией, который регулярно забывает недавние события, в то время как старые воспоминания кажутся практически нетронутыми.
Вопрос: Можете ли вы сказать мне, какой сегодня день?
Вопрос: Сколько вам лет?
Ответ: 1919.
Вопрос: Что вы ели на завтрак?
Ответ: Хлеб, картошку, пирожное …
Вопрос: Вы помните школу, в которой учились?
Ответ: школа с гуманитарным уклоном, с 20 учениками в классе, 14 мальчиков и только 6 девочек.
Первые признаки деменции:
- Изменения в процессе запоминания, которые осложняют повседневную деятельность,
- Сложности в планировании деятельности или решении задач,
- Проблемы с выполнением ежедневных обязанностей,
- Потеря ориентации во времени и пространстве,
- Трудности в понимании образов и пространственных соотношений,
- Сложности с речью и написанием слов,
- Человек оставляет вещи в странных местах или забывает свои последние действия,
- Сниженная или слабая функция суждения,
- изменения в настроении и поведении.

Не существует четкой границы между повседневными изменениями памяти и внимания и симптомами, которые должны вызвать настороженность и необходимость обратиться к врачу. Ранняя и точная диагностика болезни Альцгеймера или других форм деменции является важным шагом в выборе надлежащего лечения, ухода и поддержки.
К сожалению, до сих пор нет средства, которое могло бы вылечить или хотя бы остановить прогрессирование деменции. Определенные лекарства могут замедлить ее прогрессирование и значительно улучшить качество жизни пациентов и их опекунов. Растительный лекарственный препарат Билобил предлагает безопасное, качественное и эффективное решение для всех, кто испытывает проблемы с концентрацией внимания и памятью Больше информации о препарате Билобил.
Симптомы деменции изучены у пациентов, для которых родной язык — английский. У других все может быть иначе
Автор фото, Getty Images
Подпись к фото,Родной язык влияет на симптоматику деменции
Деменция проявляется по-разному у говорящих на английском и итальянском языках — к такому выводу пришла группа ученых Калифорнийского университета, проводившая небольшое исследование речевых проблем у пациентов с возрастными когнитивными расстройствами.
В то время как англоговорящие пациенты имели трудности в произнесении отдельных слов, пациенты с родным итальянским не испытывали такой проблемы, но говорили более простыми, короткими предложениями.
Как полагают ученые, эти выводы могут помочь точнее ставить диагноз людям разных языковых культур, поскольку в настоящий момент диагностические критерии базируются на данных, собранных в ходе наблюдений за англоговорящими пациентами.
В исследовании, проведеннном учеными Калифорнийского университета в Сан-Франциско, участвовали 20 пациентов с родным английским и 18 пациентов с родным итальянским. И у тех и у других была диагностирована первичная прогрессирующая афазия — нейродегенеративное заболевание, проявляющееся в речевых расстройствах, которые обычно сопровождают болезнь Альцгеймера и прочие формы деменции, развивающиеся преимущественно (хотя и не исключительно) в пожилом возрасте.
И у тех и у других была диагностирована первичная прогрессирующая афазия — нейродегенеративное заболевание, проявляющееся в речевых расстройствах, которые обычно сопровождают болезнь Альцгеймера и прочие формы деменции, развивающиеся преимущественно (хотя и не исключительно) в пожилом возрасте.
МРТ головного мозга и другие анализы показали схожие когнитивные функции у пациентов из обеих языковых групп. Однако, когда ученые попросили их выполнить несколько языковых упражнений, выявились различия между двумя группами.
В чем разница?
«Мы полагаем, что это происходит из-за того, что нагромождение согласных в словах, что так распространено в английском языке, представляет сложность для ухудшающихся речевых функций», — пояснила автор исследования, профессор неврологии и психиатрии Мария Луиза Горно-Темпини.
«С итальянским наоборот — легче произносить, но у него гораздо более сложная грамматика, синтаксис, в чем начинают путаться итальяноговорящие [с первичной прогрессирующей афазией]», — отметила профессор.
Таким образом, как выявило это исследование, англоговорящие пациенты имеют тенденцию меньше говорить в принципе, тогда как итальянцы не имеют проблем с произнесением слов, но проще структурируют предложения.
Английский язык относится к германской языковой группе, тогда как итальянский, наряду с французским, испанским, португальским и рядом других, принадлежит к романской, развившейся на основе латыни.
Как указывают авторы этого исследования в журнале Neurology, есть опасения, что у многих проживающих в англоязычных странах пациентов, для которых английский язык не родной, может не быть диагностирована деменция и другие нарушения мозговых функций, «поскольку их симптоматика не отвечает описанным в клинических руководствах критериям, которые основаны на изучении пациентов с родным английским».
Ученые из Калифорнийского университета намерены повторить свое исследование, но уже с большей группой разноязыких пациентов, включая пациентов с родным арабским или китайским, чтобы посмотреть на то, как проявляется симптоматика мозговых дегенеративных расстройств.
«Мы надеемся, что подобные исследования помогут нам лучше понять взаимосвязь языка и речевых расстройств, привлечь внимание к существующей разнице в лечении деменции и, в конечном счете, повсеместно улучшить медицинский уход за пациентами», — сказала профессор Горно-Темпини.
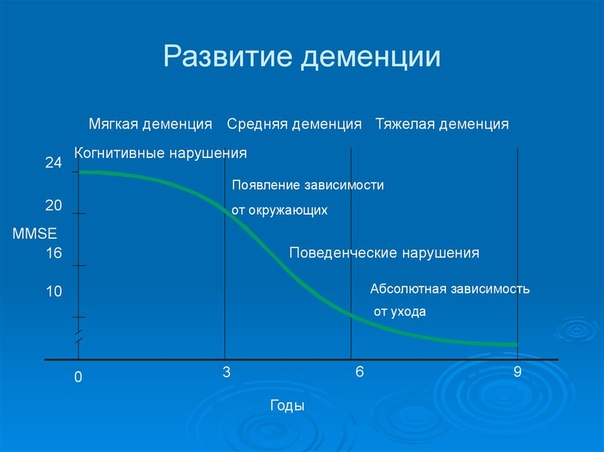
Прогрессирование деменции
{* message_confirmTraditionalInformation *} {* message_alreadyHaveAccount *}
{* #registrationForm *} {* givenName *} {* secondLastName *} {* familyName *} {* email *} {* primaryAddress_workplace *} {* mailingAddress_streetName1 *} {* mailingAddress_administrativeArea *} {* mailingAddress_municipality *} {* designation_name *} {* designation_specialty *} {* designation_specialty_json *} {* phoneNumber_mobile *} {* gender *} {* birthday *} {* traditionalRegistration_password *} {* traditionalRegistration_passwordConfirm *} {* acknowledgement_professional *} {* acknowledgement_terms *} {* hidden_programType *} {* hidden_campaign *} {* hidden_siteId *} {* hidden_preferredLanguage *} {* hidden_residencyCountry *}Натиснувши на кнопку «Підтверджую» нижче, ви погоджуєтеся з (1) можливістю обробки Ваших особистих даних компанією Pfizer, у тому числі передачу цієї інформації міжнародними каналами, відповідно до Політики конфіденційності Pfizer; (2)Політики Pfizer Cookie та Умов використання сайту компанії Pfizer.
Pfizer хотіли б розмістити cookies на вашому комп’ютері, щоб налаштувати наші пропозиції до вас через даний сайт, щоб зробити інформацію більш доступною для вас, і зробити цей сайт більш легким для подальшого користування. Щоб дізнатися більше про cookies, дивіться Політику Pfizer Cookie та Політику конфіденційності Pfizer.
Щоб дізнатися більше про cookies, дивіться Політику Pfizer Cookie та Політику конфіденційності Pfizer.
{* message_legalText *}
{*/postLoginAcceptanceForm*}БОЛЕЗНЬ АЛЬЦГЕЙМЕРА
Врач психиатр участковый диспансерного отделения
Валькова Е.Е.
Болезнь Альцгеймера — дегенеративное заболевание головного мозга, сопровождающееся образованием в мозге нейрофибриллярных клубочков и амилоидных бляшек, основным клиническим проявлением которого является прогрессирующая деменция.
В зависимости от возраста появления симптомов выделяют болезнь Альцгеймера с ранним началом (до 65 лет), составляющую от 1 до 6% всех случаев, и болезнь Альцгеймера с поздним началом (после 65 лет).
Болезнь Альцгеймера — причина развития деменции более чем в 50% случаев. В настоящее время в мире этим заболеванием страдает, по разным данным, от 24 до 36,5 млн человек. Ежегодно регистрируется более 5 млн вновь заболевших.
Наиболее значимый фактор риска болезни Альцгеймера — возраст. Вероятность развития болезни Альцгеймера возрастает с 2,8 на 1000 человек в год для людей от 65 до 69 лет до 56,1 на 1000 человек в год для лиц старше 90 лет. Приблизительно 10% людей старше 70 лет имеют признаки когнитивного снижения, и более половины из них имеют вероятную болезнь Альцгеймера. Вторым по значимости фактором риска считается отягощенный по болезни Альцгеймера семейный анамнез.
К другим факторам риска относятся женский пол, эстрогенная недостаточность у женщин, заболевания щитовидной железы, черепно-мозговые травмы, психические расстройства в анамнезе, в первую очередь — депрессия, все так называемые сосудистые факторы риска (артериальная гипертензия в среднем возрасте, нарушения углеводного и липидного обмена, заболевания сердца, нарушения мозгового кровообращения, курение).
Основное клиническое проявление болезни Альцгеймера — прогрессирующая деменция. Клиническая картина заболевания зависит от его стадии и может осложняться симптомами сопутствующих соматических и психических расстройств. Ранние симптомы болезни Альцгеймера вариабельны, чаще всего она манифестирует прогрессирующим снижением способности запоминать и воспроизводить текущую информацию, а также изменениями в поведении — появлением отстраненности, пассивности, обеднением социальных контактов и т.д.
На развернутых стадиях клиническую картину определяет собственно синдром деменции, неврологические нарушения, заострение индивидуально-психологических характеристик с последующим их нивелированием. На всех стадиях возможно развитие так называемых некогнитивных симптомов деменции — аффективных расстройств, чаще всего депрессии, бреда, галлюцинаций, а впоследствии нарушений сознания. Деменция, развивающаяся при болезни Альцгеймера, характеризуется неуклонным постепенным прогрессированием и на поздних стадиях приводит к глубоким изменениям личностных характеристик и к утрате автобиографической идентичности.
Морфофункциональные изменения при болезни Альцгеймера в наибольшей степени затрагивают гиппокамп и теменно-височные отделы коры, по мере прогрессирования заболевания в патологический процесс вовлекаются и другие зоны коры.
Около 63% пациентов с болезнью Альцгеймера переносят психотическое состояние на том или ином этапе заболевания. Около 1/3 пациентов с болезнью Альцгеймера высказывают бредовые идеи, от 11 до 30% испытывают галлюцинации, чаще слуховые и зрительные, реже — обонятельные или тактильные. Бредовые идеи наиболее часто носят персекуторный характер и часто не систематизированы. Нередко встречаются идеи ипохондрического содержания и синдром Капгра (бред двойника).
Нарушения поведения, развиваемые на поздних стадиях заболевания более чем у половины пациентов, могут быть представлены импульсивностью, вспышками вербальной или физической агрессии, расторможенностью влечений, блужданиями, аномальной двигательной активностью, психомоторным возбуждением, аутоагрессивными действиями. Хотя нарушения поведения, как правило, появляются на фоне выраженного когнитивного снижения, прямой корреляции между поведенческими и когнитивными нарушениями нет. Изменение личностных характеристик отмечается у 75% больных, оно может развиваться уже на ранних стадиях заболевания и, как правило, заключается в ригидности, апатии, сужении круга интересов, эгоцентризме, подозрительности, расторможенности, снижении критичности.
Хотя нарушения поведения, как правило, появляются на фоне выраженного когнитивного снижения, прямой корреляции между поведенческими и когнитивными нарушениями нет. Изменение личностных характеристик отмечается у 75% больных, оно может развиваться уже на ранних стадиях заболевания и, как правило, заключается в ригидности, апатии, сужении круга интересов, эгоцентризме, подозрительности, расторможенности, снижении критичности.
Для достоверного диагноза необходимо наличие следующих признаков:
— наличие синдрома деменции;
— постепенное начало с медленно нарастающим слабоумием, допускается наличие плато в развитии болезни;
— отсутствие данных в пользу обусловленности психического состояния другими системными или мозговыми заболеваниями, отсутствие острого апоплектического начала с такими неврологическими симптомами, как гемипарезы, потеря чувствительности, изменения полей зрения, нарушение координации.
Компьютерная томография и магнитно-резонансная томография позволяют исключить другие причины деменции. При болезни Альцгеймера, как правило, выявляют атрофию теменных отделов коры. Однако этот признак не является патогномоничным для болезни Альцгеймера. На ранней стадии решающее значение приобретает выявление атрофии медиальных отделов височной доли — гиппокампа и миндалины, расширения хориоидальной и гиппокамповой щели, а также височных рогов. Атрофия гиппокампа имеет прогностическую ценность и позволяет предсказать у больного с легким когнитивным снижением развитие болезни Альцгеймера.
Препаратами базисной терапии болезни Альцгеймера с доказанной эффективностью являются лекарственные средства из группы ингибиторы холинэстераз (донепезил, ривастигмин, галантамин, ипидакрин) и блокатор NMDA рецепторов (мемантин). Препараты других групп могут использоваться как средства дополнительной терапии.
Деменция — приговор? Кто в группе риска и можно ли избежать заболевания | СОВЕТЫ | ЗДОРОВЬЕ
По статистике деменцией (лат. dementia — безумие) связанной с болезнью Альцгеймера, страдает каждый девятый человек старше 65 лет, и каждый третий человек после 80 лет. Но бывает, что болезнь появляется и в более молодом возрасте.
dementia — безумие) связанной с болезнью Альцгеймера, страдает каждый девятый человек старше 65 лет, и каждый третий человек после 80 лет. Но бывает, что болезнь появляется и в более молодом возрасте.
Кто в группе риска? Можно ли избежать или отсрочить наступление опасного заболевания? Как жить с человеком, которому поставили этот диагноз, рассказала руководитель пермского Центра когнитивных нарушений Вера Черкасова.
И в 18 и в 65
Алёна Овчинникова, «АиФ-Прикамье»: В каком возрасте может развиться деменция?
Вера Черкасова: Пик расцвета когнитивных (познавательных) функций в человеческой популяции приходится на возраст 27-29 лет, затем начинается их постепенное снижение. Это естественный процесс, не являющийся заболеванием. В 30 лет наш мозг работает быстрее чем в 60. Однако появление деменции напрямую зависит от её причины: болезнь может развиться и у молодого человека, попавшего, к примеру, в аварию и получившего черепно-мозговую травму. Также причиной деменции у молодых людей могут стать токсикомании, алкоголизм, СПИД, опухоли и наследственные заболевания нервной системы, в зрелом возрасте — различные заболевания: инсульты, заболевания сердца, лёгких, нервной системы, поражения почек и печени. То есть все заболевания, прямо или косвенно поражающие головной мозг. Также признаки заболевания могут появляются и в послеоперационном периоде после перенесённого наркоза. Ну и наконец, классический пример слабоумия — болезнь Альцгеймера — участь чаще пожилых людей.
— Деменцией ведь нельзя заболеть в один день, как, например, гриппом. Какие ей предшествуют симптомы?
— У человека нарушаются память, внимание, речь, он перестаёт узнавать знакомых, не может самостоятельно правильно одеться, приготовить пищу, и выполнить любые другие действия, которые раньше не вызывали никаких затруднений – перейти улицу, выбрать нужный транспорт, купить необходимые продукты. Специалисты говорят – «пациент с деменцией не справляется с жизнью». Иными словами, наступает стадия бытовой дезадаптации. Да, заболевание развивается постепенно, на самых ранних стадиях человек может работать, выполняя свои профессиональные обязанности, если они не требуют быстрой реакции и принятия сложных решений. При когнитивных нарушениях у человека пропадает способность к пониманию эмоций и логики других людей, он не может правильно поставить себе цель своей деятельности: для чего я это делаю? Порой выполняет бессмысленные действия.
Специалисты говорят – «пациент с деменцией не справляется с жизнью». Иными словами, наступает стадия бытовой дезадаптации. Да, заболевание развивается постепенно, на самых ранних стадиях человек может работать, выполняя свои профессиональные обязанности, если они не требуют быстрой реакции и принятия сложных решений. При когнитивных нарушениях у человека пропадает способность к пониманию эмоций и логики других людей, он не может правильно поставить себе цель своей деятельности: для чего я это делаю? Порой выполняет бессмысленные действия.
— Может ли человек, находясь в здравом ещё уме заметить у себя эти нарушения и вылечить болезнь на начальной стадии?
— Вот как чаще всего складывается ситуация: на начальных стадиях, если это не остро развившаяся деменция, человек замечает, что начинает сдавать, но стыдится дефекта и скрывает его. Примерно в половине случаев лёгкие или умеренные когнитивные нарушения переходят в тяжёлые и человек просто перестаёт их замечать. Как говорят по этому поводу специалисты, «слабость рассудка не позволяет заметить его слабость». И если у пожилого человека нет жалоб на забывчивость, то с большой долей вероятности у него уже имеется деменция. А ведь врачи в первую очередь ориентированы на жалобы пациентов. Это очень важный момент! Пациент с этим заболеванием, имея уже серьёзные сложности с простыми бытовыми навыками, жалуется на что угодно, но не на снижение памяти. Родственникам таких больных приходится очень тяжело. Поэтому, чем раньше задуматься о профилактике и лечении, тем меньше проблем будет с пожилыми родственниками в будущем.
Спорт, питание, сон
— Ещё обладая критикой своего состояния, может ли человек с начинающейся деменцией улучшить своё будущее: пить витамины, делать определённые упражнения? Можно ли отсрочить наступление болезни?
— Самолечением заниматься опасно. Биологические активные добавки – это не лекарства и они не лечат. Витамины тоже небезопасны. Многие из них не сочетаются и друг с другом и с теми лекарствами, которые принимает человек, а также имеют противопоказания при различных заболеваниях. Чтобы уменьшить риск развития деменции в целом и болезни Альцгеймера в частности, разработаны современные рекомендации: это физическая активность (ходьба, северная ходьба, плавание, катание на велосипеде, на лыжах и другие), правильное питание и режим сна. Важно употреблять в пищу много клетчатки: каши, овощи и фрукты. Именно с помощью клетчатки выводятся из организма токсины и поступают витамины и микроэлементы. Также нужно придерживаться средиземноморской диеты: употреблять оливковое масло, больше рыбы, чем мяса. Из напитков — зелёный чай, натуральный кофе, допускаются сухие вина. А ещё продукты, богатые витамином С: чёрная смородина, лимон, киви, приправа кари. И никакого ночного просмотра телевизора или игр на компьютере. Ночью у нас вырабатывается гормон молодости мелатонин, который замедляет старение головного мозга.
— Реально ли вылечить человека от этой болезни?
— Большая часть когнитивных нарушений, связанных с поражением нейрона, необратимы. По какой причине возникает болезнь Альгеймера, тоже пока неизвестно. Те медикаменты, которые назначают при деменции не вылечивают болезнь полностью, но оказывают выраженный симптоматический эффект и замедляют прогрессирование заболевания. Очень важно вовремя начать правильную терапию.
— Деменция мужская или женская проблема?
— У женщин и мужчин разные виды деменции. Например, лобно-височную деменцию (страдает в первую очередь поведение, люди ведут себя не адекватно, проявляют агрессию и апатию), а также диффузное поражение головного мозга чаще обнаруживают у мужчин. Черепно-мозговые травмы, инсульты — тоже чаще бывают у мужчин.
Выделяют две формы болезни Альцгеймера: раннюю и позднюю. Первая встречается реже, но протекает очень тяжело. Начинается в возрасте 40+, чаще у женщин. Как правило страдают ей люди с высшим образованием, часто занимающие руководящие посты. К примеру, на днях у меня на приёме была женщина, в недалеком прошлом руководитель. Её привела подруга, которая не смогла равнодушно смотреть на разрушение личности близкого человека. У таких пациентов быстро ухудшается состояние: за несколько лет они теряют память, не могут посчитать деньги и расплатиться в магазине, не в силах определить, на каком транспорте им ехать до дома, забывают, как зовут детей, родственников, как пользоваться повседневными приборами — телефоном, компьютером, микроволновкой и т.д. Деменция быстро прогрессирует. И пока критика ещё сохраняется, человек чувствует, что он очень сильно сдал, но изменить что-либо не в силах.
Как правило страдают ей люди с высшим образованием, часто занимающие руководящие посты. К примеру, на днях у меня на приёме была женщина, в недалеком прошлом руководитель. Её привела подруга, которая не смогла равнодушно смотреть на разрушение личности близкого человека. У таких пациентов быстро ухудшается состояние: за несколько лет они теряют память, не могут посчитать деньги и расплатиться в магазине, не в силах определить, на каком транспорте им ехать до дома, забывают, как зовут детей, родственников, как пользоваться повседневными приборами — телефоном, компьютером, микроволновкой и т.д. Деменция быстро прогрессирует. И пока критика ещё сохраняется, человек чувствует, что он очень сильно сдал, но изменить что-либо не в силах.
К чему есть интерес?
— Наверняка, на прогрессирующее слабоумие родителей чаще жалуются их дети. К каким специалистам нужно идти?
— Детям важно понять, что причина состояния родителя «не в себе» не случайна. Не потому, что он не выспался или находится в плохом настроении. Если пожилые родители не справляются с обычными бытовыми функциями, это ненормально, это болезнь. Если человек не может приготовить еду, воспользоваться телефоном, одеться, значит нужно идти к врачу. Задача доктора — поставить диагноз и назначить лечение. Это делают невролог, гериатр. На поздних стадиях нужно сразу обращаться к психиатру. В сложных случаях целесообразна консультация в краевом Центре когнитивных нарушений. На бесплатный приём к нам можно записаться с направлением от участкового терапевта.
— Как правильно вести себя с заболевшим родственником?
— Если в семье живёт человек с деменцией, от этого в первую очередь страдают его окружающие. На поздних стадиях пациент теряет критику к своему состоянию и может не просто совершать бессмысленные действия, но и представлять опасность для окружающих: включить газ, воду, затопить соседей, уйти из дома. Часто бывает, что родственники, желая тренировать больного, заставляют его выучить стихотворение или разгадать кроссворд. Это не имеет абсолютно никакого смысла и порождает конфликты. Да, когнитивная тренировка нужна, она проводится либо под контролем нейропсихолога, либо в домашних условиях. Больной человек должен заниматься такой активностью, которая ему интересна, приятна и доступна. Что это может быть? Занятие надо выбирать индивидуально, опираясь на увлечения, интересы, сохранённые функции и профессиональные навыки. Если бабушка всю жизнь вязала, то пусть продолжает, но в более простом варианте. Если двигательные функции сохранились и зрение позволяет, то пусть человек играет в шахматы, домино, любые игры, не вызывающие неприятия. Важно, чтобы он испытывал при этом положительные эмоции. Ну и конечно, таким пациентам обязательна ежедневная физическая активность: стандартные 10 тысяч шагов в день, утренняя зарядка, в том числе и силовые упражнения.
Это не имеет абсолютно никакого смысла и порождает конфликты. Да, когнитивная тренировка нужна, она проводится либо под контролем нейропсихолога, либо в домашних условиях. Больной человек должен заниматься такой активностью, которая ему интересна, приятна и доступна. Что это может быть? Занятие надо выбирать индивидуально, опираясь на увлечения, интересы, сохранённые функции и профессиональные навыки. Если бабушка всю жизнь вязала, то пусть продолжает, но в более простом варианте. Если двигательные функции сохранились и зрение позволяет, то пусть человек играет в шахматы, домино, любые игры, не вызывающие неприятия. Важно, чтобы он испытывал при этом положительные эмоции. Ну и конечно, таким пациентам обязательна ежедневная физическая активность: стандартные 10 тысяч шагов в день, утренняя зарядка, в том числе и силовые упражнения.
— В России пенсионный возраст повысили. Значит ли это, что обслуживать нас или управлять нами будут люди с деменцией?
— Да, вполне вероятно. И мы не дооцениваем эту опасность. Представляете, как будет управлять поездом машинист, страдающий деменцией? Остаётся надеяться, что проводимые медицинские осмотры смогут выявить у лиц опасных профессий заболевания, сопровождающиеся развитием деменции.
Вера Черкасова, родилась 9 декабря 1970 года в Перми. В 1994 году с отличием закончила лечебный факультет ПГМА. Затем работала невропатологом в Березниковской клинической больнице. С 2009 года – зав. кафедрой медицинской реабилитации и спортивной медицины в ПГМУ, с 2018 года – директор учебно-методического Центра нейрогериатрии и дементологии в госпитале ветеранов войн.Клинический случай прогрессирующей сосудистой лейкоэнцефалопатии | Федулаев
1. Blass JP, Hoyer S, Nitsch R. A translation of Otto Binswanger’s article ‘The delineation of the generalized progressive paralyses’. 1894. Archives of neurology. 1991; 48 (9): 961–972.
1991; 48 (9): 961–972.
2. Caplan LR. Binswanger’s disease — revisited. Neurology. 1995; 45 (4): 626–633.
3. Jonsson M, Zetterberg H, van Straaten E, et al. Cerebrospinal fluid biomarkers of white matter lesions — cross-sectional results from the LADIS study. Eur J Neurol. 2010; 17 (3): 377–382.
4. Менделевич Д. М., Сафина Г. Д. Психические расстройства при болезни Бинсвангера. // Неврологический вестник. — 2003. — Т. XXXV, вып. 1–2. — С. 64–67.
5. Никифоров А. С., Коновалов А. Н., Гусев Е. И. Клиническая неврология. В 3 томах. М.: Медицина, 2002: 548–552.
6. Bennett D. A., Wilson R. S., Gilley D. W., Fox J. H. Clinical diagnosis of Binswanger’s disease. // Journal of Neurology, Neurosurgery & Psychiatry. 1990; 53: 961–965. doi.org/l0.1136/jnnp.53.11.961.
7. Медведев А. В., Корсакова Н. К., Саватеева Н. Ю. О деменции при энцефалопатии Бинсвангера. // Клиническая геронтология. 1996; 2: 17–28.
8. Кадыков А. С. Дисциркуляторная энцефалопатия: алгоритм диагностики и лечения у больных с артериальной гипертензией. / А. С. Кадыков, Н. В. Шахпаронова // Неврология, нейропсихиатрия, психосоматика. — 2010. — № 3. — С. 12–27.
9. Hachinski V. Binswanger’s disease: neither Binswanger’s nor a disease. // Journal of the Neurological Sciences. 1991; 103 (1). doi. org/10.1016/0022–510x(91)90274-b.
10. Солошенкова О. О., Чукаева И. И., Орлова Н. В. / Дислипидемии в клинической практике. Часть 1. Лечебное дело. 2009. № 3. С. 12–17.
Солошенкова О. О., Чукаева И. И., Орлова Н. В. / Дислипидемии в клинической практике. Часть 1. Лечебное дело. 2009. № 3. С. 12–17.
11. Орлова Н. В. Воспаление и факторы риска сердечно-сосудистых заболеваний. Диссертация на соискание ученой степени доктора медицинских наук. / ГОУВПО «Российский государственный медицинский университет». Москва, 2008.
Обследование по быстро прогрессирующей деменции | Центр памяти и старения
При подозрении на возможное быстро прогрессирующее слабоумие (РПЗ) вам следует заказать определенные тесты, чтобы исключить или подтвердить диагноз. Для полноценного обследования необходимо множество тестов, но МРТ головного мозга, включая FLAIR (восстановление с инверсией с ослаблением жидкости) и DWI (диффузионно-взвешенная визуализация), является единственным наиболее полезным инструментом в диагностике CJD.
Медицинский осмотр и история болезни
Поскольку первые симптомы CJD иногда носят конституциональный характер, определение фактической даты начала может потребовать некоторого исследования.Оценка способности пациента выполнять повседневную деятельность помогает количественно оценить прогрессирование заболевания. Спросите о семейном анамнезе неврологических или психических заболеваний (родственникам, возможно, поставили неправильный диагноз; многих пациентов с генетическим прионным заболеванием ошибочно принимают за другие неврологические или психические расстройства, включая болезнь Альцгеймера, болезнь Паркинсона или атипичное паркинсоническое деменцию). Медицинский анамнез и история путешествий могут помочь оценить риск приобретенного CJD.
Работа с кровью
Следующие лаборатории охватят обширную основу для начального обследования: общий анализ крови, химическая панель (включая Ca, Mg и P), LFT, RPR, ревматологический скрининг (СОЭ, ANA, RF, CRP, C-ANCA и P-ANCA) , функция щитовидной железы, B12, гомоцистеин, антитела против тиреоглобулина и тиреопероксидазы, ВИЧ, антитела Лайма и паранеопластические антитела. Мы рекомендуем анализировать кровь на генетические формы прионной болезни у всех пациентов с подозрением на БКЯ, особенно у пациентов с семейным анамнезом деменции, мозжечкового или паркинсонического расстройства. У более чем 60% пациентов, у которых выявлено генетическое прионное заболевание, семейный анамнез прионного заболевания неизвестен. См. Раздел «Дифференциальная диагностика CJD» для дополнительных предлагаемых тестов.
Мы рекомендуем анализировать кровь на генетические формы прионной болезни у всех пациентов с подозрением на БКЯ, особенно у пациентов с семейным анамнезом деменции, мозжечкового или паркинсонического расстройства. У более чем 60% пациентов, у которых выявлено генетическое прионное заболевание, семейный анамнез прионного заболевания неизвестен. См. Раздел «Дифференциальная диагностика CJD» для дополнительных предлагаемых тестов.
Общий анализ мочи
Для исключения инфекции необходимо отправитьUA и посевы. Соберите суточную мочу для проверки на наличие тяжелых металлов, включая мышьяк, свинец, ртуть, медь, алюминий и висмут, с историей воздействия.
МРТ
Это единственный самый полезный тест , который вы можете сделать для диагностики CJD. Закажите аксиальную и корональную последовательности T1, T2, DWI (и карту ADC) и FLAIR. Чтобы исключить другие состояния, необходимо провести хотя бы одно МРТ с контрастом и без него.
ЭЭГ
У большинства людей с БКЯ ЭЭГ покажет неспецифическое замедление активности, которое наблюдается при многих формах деменции. Около 65% пациентов с sCJD будут демонстрировать характерные периодические комплексы резких волн (PSWC) примерно раз в секунду, но часто только на поздних стадиях течения болезни.Эти изменения также не характерны для CJD; они могут возникать при печеночной энцефалопатии, энцефалопатии Хашимото и поздних стадиях других нейродегенеративных заболеваний, таких как болезнь Альцгеймера (AD) и деменция с тельцами Леви (DLB).
CT
КТ грудной клетки, брюшной полости и таза с внутривенным контрастированием и без него может помочь исключить злокачественные новообразования и паранеопластические заболевания.
Биопсия головного мозга
Используя DWI MRI, мы редко делаем биопсию головного мозга. Однако биопсия головного мозга иногда может помочь подтвердить CJD или другой диагноз.Биопсия головного мозга, даже при БКЯ, не может быть диагностической, так как не всегда берут ткань из пораженной части мозга.
прионных болезней | Центр памяти и старения
Прионные болезни вызываются аномальными прионами, микроскопическими инфекционными агентами, состоящими из белка. Прионы вызывают ряд заболеваний у различных млекопитающих, включая губчатую энцефалопатию крупного рогатого скота (коровье бешенство или ) у крупного рогатого скота и скрейпи у овец.
Существует три различных подтипа прионной болезни, разделенных на категории в зависимости от того, как болезнь передается. Все они немного различаются по типичным признакам, симптомам и продолжительности болезни. Подтипы спорадические, генетические и приобретенные.
Спорадические прионные болезни
- Спорадическая болезнь Крейтцфельда-Якоба (sCJD)
Причина «классической» или «спорадической» CJD неизвестна, что означает, что она возникает у людей без каких-либо известных факторов риска или генных мутаций.Типичные симптомы включают дисбаланс и нарушение координации, потерю памяти и нарушение мышления, а также психиатрические симптомы, такие как тревога или депрессия. Как только симптомы действительно появляются, CJD очень быстро прогрессирует и обычно заканчивается летальным исходом в течение нескольких месяцев после появления симптомов. sCJD обычно поражает людей в возрасте от 60 лет и редко встречается у людей моложе 40 лет. Спорадическая CJD — наиболее распространенная форма. - Спорадическая фатальная бессонница (sFI)
Подобно sCJD, sFI вызывается неправильно свернутым белком, но также, как sCJD, причина неправильного свертывания этого белка неизвестна.Симптомы включают трудности с засыпанием (бессонницу), трудности при ходьбе, потерю веса и чрезмерные слезы на глазах. Симптомы быстро прогрессируют до потери сознания и смерти.
 Симптомы быстро прогрессируют до потери сознания и смерти.
Симптомы быстро прогрессируют до потери сознания и смерти.Генетические прионные болезни
- Семейная болезнь Крейтцфельда-Якоба (fCJD)
Унаследованные мутации в гене прионного белка ( PRNP ) вызывают семейную форму прионной болезни. Этот прионный ген предоставляет вашим клеткам инструкции относительно того, как производить прионный белок.При fCJD мутации в этом гене заставляют клетки продуцировать аномальную форму прионного белка вместо нормальной формы. В большинстве случаев пациент с fCJD наследует измененный ген от одного больного родителя. У некоторых людей новая мутация в гене вызывает fCJD. Хотя у таких людей, скорее всего, нет пострадавшего родителя, они могут передать генетическое изменение своим детям. Симптомы зависят от типа мутации, но часто выглядят как классический CJD: проблемы с балансом и координацией, потеря памяти и нарушение мышления.Обычно эти симптомы появляются раньше при fCJD, чем при sCJD. Продолжительность болезни обычно больше, чем при спорадической форме. На генетически унаследованный подтип приходится около 15% случаев CJD. - Синдром Герстмана-Штраусслера-Шейнкера (GSS)
Болезнь Герстмана-Штрауслера-Шейнкера (GSS) — чрезвычайно редкое нейродегенеративное заболевание головного мозга. Он почти всегда передается по наследству и встречается только в нескольких семьях по всему миру. Заболевание обычно возникает в возрасте от 35 до 55 лет.На ранних стадиях пациенты могут испытывать атаксию различной степени (отсутствие мышечной координации), включая неуклюжесть, неустойчивость и трудности при ходьбе. По мере прогрессирования заболевания атаксия становится более выраженной, и у большинства пациентов развивается деменция. Другие симптомы могут включать дизартрию (невнятность речи), нистагм (непроизвольные движения глаз), спастичность (жесткий мышечный тонус) и нарушения зрения, иногда приводящие к слепоте. Также может возникнуть глухота. В некоторых семьях присутствуют признаки паркинсонизма. Он наследуется по аутосомно-доминантному типу из-за изменения гена приона на хромосоме 20pter-p12. - Смертельная семейная бессонница (FFI)
Смертельная семейная бессонница (FFI) — очень редкое аутосомно-доминантное наследственное заболевание головного мозга. Ответственный за это доминантный ген был обнаружен всего в 28 семьях по всему миру; если ген есть только у одного из родителей, потомство имеет 50% шанс унаследовать его и заболеть. Переход пациента к полной бессоннице неизлечим и в конечном итоге фатален.Выделяют четыре стадии болезни. Первая стадия — прогрессирующая бессонница, признак фатальной семейной бессонницы. Первая стадия длится примерно четыре месяца и включает в себя набор психиатрических проблем, таких как панические атаки и причудливые фобии. Вторая стадия включает галлюцинации, панику, возбуждение и потливость и длится около пяти месяцев. Третий этап длится около трех месяцев и представляет собой полную бессонницу с похуданием. В этот момент человек выглядит намного старше и может испытывать недержание мочи.Четвертая стадия длится около шести месяцев и распознается как деменция, полная бессонница и внезапная смерть после потери речи.
 Он наследуется по аутосомно-доминантному типу из-за изменения гена приона на хромосоме 20pter-p12.
Он наследуется по аутосомно-доминантному типу из-за изменения гена приона на хромосоме 20pter-p12.Приобретенные прионные болезни
CJD, приобретенный в результате воздействия аномального прионного белка, составляет менее 1% известных случаев CJD . Важно понимать, что CJD, вызванный воздействием приона, очень редок. Есть три подтипа этой формы:
- Ятрогенная болезнь Крейтцфельдта-Якоба (iCJD)
«Ятрогенная» буквально означает «вызванная врачом».«Таким образом, ятрогенное заболевание — это заболевание, связанное с врачом или лечением. Признаки и симптомы часто напоминают классический CJD. Возраст начала заболевания зависит от возраста экспозиции и времени инкубации. Число новых случаев iCJD резко сократилось, поскольку практика предотвращения заражения изменилась. Случаи CJD были связаны с лечением с использованием гормона роста, полученного из гипофиза человека. К счастью, в 1980-х годах была разработана синтетическая версия гормона роста человека, поэтому теперь гормон роста создается в лаборатории, а не у людей.Несколько случаев CJD были связаны с трансплантатами инфицированных тканей и трансплантатами от доноров, у которых оказалось, что CJD. Нет известных случаев, когда спорадическая или семейная форма CJD передавалась другим людям при переливании крови. Несколько случаев типа CJD, связанного с употреблением в пищу инфицированной говядины, передавались через переливание крови (см. Описание ниже). Доноры органов, тканей и крови теперь проходят скрининг на факторы риска БКЯ, и им не разрешается сдавать кровь, если они потенциально могут передать аномальные прионы реципиенту.Также было несколько случаев, связанных с зараженными инструментами, используемыми в хирургии головного мозга. Поскольку типичные процедуры стерилизации не устраняют аномальные прионы, текущие рекомендации состоят в том, чтобы уничтожить инструменты, которые использовались у пациента с CJD или с подозрением на CJD. - Вариант болезни Крейтцфельда-Якоба (vCJD)
Эта форма была связана с употреблением в пищу говядины, зараженной губчатой энцефалопатией крупного рогатого скота (BSE или «коровье бешенство») у крупного рогатого скота. На ранних стадиях у пациентов часто наблюдаются изменения личности и психиатрические симптомы, такие как депрессия или ломка.Психиатрические симптомы часто являются наиболее заметным признаком ранней болезни БКЯ, но деменция развивается позже. Двигательные симптомы vCJD (спотыкание, падения и затруднения при ходьбе) также имеют тенденцию проявляться раньше при vCJD, чем при классической CJD. Расчетный инкубационный период составляет от 5 до 40 лет, а продолжительность болезни обычно составляет 12–14 месяцев после появления признаков и симптомов. vCJD поражает людей в возрасте от 20 до 20 лет, что намного раньше, чем у людей со спорадической CJD. Один человек с вариантом CJD был идентифицирован в Соединенных Штатах и один в Канаде, однако оба жили в Великобритании во время эпидемии BSE и заразились от заражения в Великобритании. - Куру
В 1950–1960-х годах куру достигло масштабов эпидемии в племени Саут-Форе в Папуа-Новой Гвинее. Хотя исследователи не знают, как это началось, они знают, что это распространилось, когда члены племени ритуально потребляли ткани пострадавших людей во время погребальных обрядов. Куру характеризуется проблемами при ходьбе, дрожанием конечностей, невнятной речью и изменениями настроения, но незначительным слабоумием или его отсутствием. Обычно это приводит к летальному исходу в течение 6–12 месяцев. Куру исчез с прекращением каннибалистических практик в Новой Гвинее.
 К счастью, в 1980-х годах была разработана синтетическая версия гормона роста человека, поэтому теперь гормон роста создается в лаборатории, а не у людей.Несколько случаев CJD были связаны с трансплантатами инфицированных тканей и трансплантатами от доноров, у которых оказалось, что CJD. Нет известных случаев, когда спорадическая или семейная форма CJD передавалась другим людям при переливании крови. Несколько случаев типа CJD, связанного с употреблением в пищу инфицированной говядины, передавались через переливание крови (см. Описание ниже). Доноры органов, тканей и крови теперь проходят скрининг на факторы риска БКЯ, и им не разрешается сдавать кровь, если они потенциально могут передать аномальные прионы реципиенту.Также было несколько случаев, связанных с зараженными инструментами, используемыми в хирургии головного мозга. Поскольку типичные процедуры стерилизации не устраняют аномальные прионы, текущие рекомендации состоят в том, чтобы уничтожить инструменты, которые использовались у пациента с CJD или с подозрением на CJD.
К счастью, в 1980-х годах была разработана синтетическая версия гормона роста человека, поэтому теперь гормон роста создается в лаборатории, а не у людей.Несколько случаев CJD были связаны с трансплантатами инфицированных тканей и трансплантатами от доноров, у которых оказалось, что CJD. Нет известных случаев, когда спорадическая или семейная форма CJD передавалась другим людям при переливании крови. Несколько случаев типа CJD, связанного с употреблением в пищу инфицированной говядины, передавались через переливание крови (см. Описание ниже). Доноры органов, тканей и крови теперь проходят скрининг на факторы риска БКЯ, и им не разрешается сдавать кровь, если они потенциально могут передать аномальные прионы реципиенту.Также было несколько случаев, связанных с зараженными инструментами, используемыми в хирургии головного мозга. Поскольку типичные процедуры стерилизации не устраняют аномальные прионы, текущие рекомендации состоят в том, чтобы уничтожить инструменты, которые использовались у пациента с CJD или с подозрением на CJD.
Центр FTD в Пенсильвании | Быстро прогрессирующая деменция
ОбзорБольшинство нейродегенеративных заболеваний, таких как лобно-височная дегенерация или болезнь Альцгеймера, прогрессируют медленно, постепенно в течение нескольких лет. Однако у некоторых пациентов когнитивные, моторные или поведенческие симптомы могут проявляться и прогрессировать в течение недель или месяцев. Эти состояния в совокупности называются «быстро прогрессирующими деменциями».”
Этиология Существует несколько этиологий, которые потенциально могут вызвать подострое неврологическое снижение «быстро прогрессирующей деменции». Во-первых, это осложнения заболеваний, которые могут вызвать подострый спад, включая метаболические, инфекционные, воспалительные, опухолевые или сосудистые состояния. Каждая из этих категорий заболеваний имеет определенные стратегии лечения, которые важно определить на ранней стадии развития болезни. Примеры заболеваний, которые могут вызвать быстрое снижение познавательной способности, включают аномалии щитовидной железы, аутоиммунные заболевания (т.е. иммунная система организма атакует сама себя, как при системной красной волчанке), инфекции нервной системы (например, менингит), сердечную или легочную недостаточность. Во-вторых, существуют состояния, которые воздействуют на центральную нервную систему. Примеры включают нейродегенеративное состояние, подострую губчатую энцефалопатию или болезнь Крейтцфельдта-Якоба, аутоиммунные заболевания центральной нервной системы (то есть расстройство анти-NMDA-рецептор-антитело или лимбический энцефалит) и определенные типы судорог (т.е. частичный комплексный эпилептический статус). Общие симптомы включают спутанность сознания, потерю памяти, нарушение равновесия, судороги или ригидность; однако симптомы могут сильно различаться у разных людей и часто различаются в зависимости от основной причины состояния. Диагностика этих состояний включает тщательное неврологическое обследование и сбор анамнеза вместе с систематическим исследованием серологических, нейровизуализационных и других подтверждающих тестов. Penn Frontotemporal Degeneration Center активно оценивает пациентов для выявления этих ненейродегенеративных состояний, чтобы можно было начать соответствующее лечение.
Примеры включают нейродегенеративное состояние, подострую губчатую энцефалопатию или болезнь Крейтцфельдта-Якоба, аутоиммунные заболевания центральной нервной системы (то есть расстройство анти-NMDA-рецептор-антитело или лимбический энцефалит) и определенные типы судорог (т.е. частичный комплексный эпилептический статус). Общие симптомы включают спутанность сознания, потерю памяти, нарушение равновесия, судороги или ригидность; однако симптомы могут сильно различаться у разных людей и часто различаются в зависимости от основной причины состояния. Диагностика этих состояний включает тщательное неврологическое обследование и сбор анамнеза вместе с систематическим исследованием серологических, нейровизуализационных и других подтверждающих тестов. Penn Frontotemporal Degeneration Center активно оценивает пациентов для выявления этих ненейродегенеративных состояний, чтобы можно было начать соответствующее лечение.
Быстро прогрессирующая нейродегенеративная деменция | Деменция и когнитивные нарушения | JAMA Neurology
Фон Нейродегенеративная деменция обычно характеризуется незаметным началом и относительно медленно прогрессирующим течением. Реже встречаются пациенты с быстро прогрессирующим течением до смерти.
Объектив Для характеристики пациентов с нейродегенеративным заболеванием и быстро прогрессирующим течением до смерти.
Дизайн, условия и пациенты Используя поиск по тексту для слов «быстрое» и «слабоумие» в одном предложении, система связи медицинских записей клиники Мэйо использовалась для идентификации всех пациентов, обследованных в период с 1 января 2000 г. по 30 сентября 2007 г. с аутопсией головного мозга (N = 96) в медицинском центре третичного уровня. Из этих 96 пациентов мы включили только тех, у которых продолжительность заболевания менее 4 лет до смерти и с гистологическим диагнозом нейродегенеративного заболевания.
Основные показатели результатов Быстро прогрессирующее слабоумие со смертью раньше, чем через 4 года после возникновения и патологического диагноза в нашем учреждении нейродегенеративного заболевания.
Результаты Мы включили 22 пациента (10 мужчин). Хотя 8 случаев (36%) имели болезнь Крейтцфельда-Якоба (БКЯ), в остальных случаях наблюдалась лобно-височная долевая дегенерация с дегенерацией двигательных нейронов (5 случаев [23%]), таупатия (прогрессирующий надъядерный паралич или кортикобазальная дегенерация) (4 случая [18]. %]), диффузной болезни с тельцами Леви (3 случая [14%]) или болезни Альцгеймера (2 случая [9%]).Все пациенты с CJD умерли через 12 месяцев или раньше после начала болезни, в то время как у остальных болезнь продолжалась более 12 месяцев. Примечательно, что все 3 пациента с диффузной болезнью с тельцами Леви, но никто из других, первоначально не испытали преходящую послеоперационную или связанную с заболеванием энцефалопатию, затем относительную нормальность в течение 2 лет и, наконец, быстро прогрессирующую деменцию и снижение смертности в течение 4-12 месяцев.
Выводы Основываясь на этой когорте, хотя CJD является наиболее вероятной причиной быстро прогрессирующей нейродегенеративной деменции, лобно-височная долевая дегенерация с дегенерацией моторных нейронов, диффузная болезнь с тельцами Леви, таупатии и болезнь Альцгеймера также могут вызывать быстро прогрессирующую деменцию.Если продолжительность болезни превышает 12 месяцев, нейродегенеративное заболевание, не связанное с CJD, может быть более вероятным, чем CJD.
Термин деменция означает наличие когнитивных и / или поведенческих нарушений, которые существенно влияют на повседневную деятельность человека. 1 Однако это общий термин, охватывающий множество различных этиологий, включая нейродегенеративные, метаболические, сосудистые и инфекционные заболевания.
Отдельные нейродегенеративные заболевания имеют относительно специфические клинические признаки 2 и гистологически характеризуются различной степенью потери нейронов, глиозом и обычно аномальным отложением белка.Гистологические особенности в конечном итоге определяют каждое нейродегенеративное заболевание. Наиболее распространенными нейродегенеративными расстройствами, сопровождающимися деменцией, являются болезнь Альцгеймера (AD), диффузная болезнь с тельцами Леви (DLBD) и лобно-височная долевая дегенерация (FTLD). В этих условиях деменция обычно коварна в начале, очень медленно прогрессирует, а общая продолжительность болезни превышает 5 лет. 3 , 4
Более быстро прогрессирующая деменция наблюдалась при прионных заболеваниях, особенно при болезни Крейтцфельдта-Якоба (CJD), 5 , при которой продолжительность болезни обычно составляет менее 1 года.Однако время от времени мы сталкивались с быстро прогрессирующими, в конечном итоге фатальными деменциями с клиническими признаками, типичными для других нейродегенеративных заболеваний, за исключением временного течения. Таким образом, целью данного исследования было охарактеризовать подтвержденные аутопсией случаи нейродегенеративных заболеваний с быстро прогрессирующей деменцией и сравнить их с подтвержденными аутопсией случаями CJD.
Используя поиск по тексту для слов «быстрое» и «слабоумие» в одном предложении, система связи медицинских записей клиники Мэйо использовалась для выявления всех пациентов с возможным быстро прогрессирующим слабоумием, которые проходили обследование в нашем учреждении в период с 1 января 2000 г. 30 сентября 2007 г. прошла вскрытие мозга в нашем учреждении, поставила патологический диагноз.Всего выявлено 96 случаев. Медицинские карты 96 случаев были просмотрены нейродегенеративным специалистом (K.A.J.), не имеющим отношения к диагнозу невропатологии. Из этих 96 случаев в исследование были включены только пациенты с общей продолжительностью заболевания менее 4 лет до смерти и гистологически подтвержденным диагнозом нейродегенеративного заболевания. Случаи, которые прошли патологическое обследование в другом месте с патологическим отчетом только по почте или факсу, были исключены (n = 3).
Из этих 96 случаев в исследование были включены только пациенты с общей продолжительностью заболевания менее 4 лет до смерти и гистологически подтвержденным диагнозом нейродегенеративного заболевания. Случаи, которые прошли патологическое обследование в другом месте с патологическим отчетом только по почте или факсу, были исключены (n = 3).
Клинические данные были извлечены по всем случаям, которые соответствовали нашим критериям включения и исключения, включая демографические особенности, первые клинические признаки, а также первоначальный и окончательный диагнозы до смерти.Начало заболевания определялось как месяц, в котором семья пациента заметила первый неврологический симптом, который мог быть связан с дегенеративным процессом. Наличие или отсутствие нарушений поведения во сне, связанных с быстрым движением глаз, серьезные колебания, 6 паркинсонизм (≥2 из следующего: тремор, брадикинезия, ригидность или постуральная нестабильность), галлюцинации и бред, а также заболевание двигательных нейронов, а также результаты лабораторных исследований спинномозговой жидкости (ЦСЖ), электроэнцефалографии (ЭЭГ) и нейровизуализации.Расстройство поведения во сне с быстрым движением глаз считалось положительным, если поведение соответствовало диагностическим критериям В для расстройства поведения во сне с быстрым движением глаз, определяемого как ненормальное, дикое рывковое движение, возникающее во время сна, с травмами, связанными со сном, или движениями, которые потенциально вредны или разрушительны. 7 Принимая во внимание недавние сообщения об ассоциации антитиропероксидазы, антимикросомных и паранеопластических антител с быстро прогрессирующей деменцией, были также зарегистрированы уровни антител к щитовидной железе и результаты паранеопластических тестов.
Невропатологическое обследование
Невропатологическое обследование было проведено одним из двух сертифицированных невропатологов (J. E.P. и C.G.), которые не ослепляли клинический диагноз. Консенсус между обоими патологами был достигнут в том случае, если случай был сложным (n = 1). Во всех случаях мы брали образцы лобной, височной, теменной и затылочной коры, миндалины, гиппокампа и поясной извилины, базального ядра, базальных ганглиев, таламуса, среднего мозга, моста, продолговатого мозга и мозжечка.Все пациенты прошли гистологическое исследование гематоксилин-эозином, а также модифицированное окрашивание серебром по Бельшовскому. Иммуногистохимия с антителами к тау (клон AT8 с титром 1: 3750; Endogen, Woburn, Массачусетс), β-амилоид (клон 6F3D с титром 1:20; Novocastra Laboratories, Ньюкасл-апон-Тайн, Англия), α-синуклеин (клон LB509 с титром 1:25; Zymed Laboratories, Inc, Южный Сан-Франциско, Калифорния), убиквитин (поликлональный с титром 1: 100; Dako North America, Inc, Карпинтерия, Калифорния), нейрофиламент (клон 2F11 с титр 1: 800; Dako North America, Inc), TDP-43 (с титром 1: 2000; ProteinTech Group, Inc, Чикаго, Иллинойс) и прионный белок (клон 3F4 с титром 1:50; Dako North America, Inc).Все патологические диагнозы были поставлены на основании ранее опубликованных критериев. 8 -14 Особое внимание уделялось наличию любых других патологических проявлений, включая вторичные нейродегенеративные заболевания (TDP-43-положительные включения, тельца Леви, патологические изменения типа Альцгеймера), амилоидную ангиопатию, сосудистые патологические признаки любых сорт, и аргирофильная болезнь зерна. Все случаи CJD были подтверждены вестерн-блоттингом на наличие прионного белка в Национальном центре наблюдения за патологией прионных заболеваний, Кливленд, Огайо.
E.P. и C.G.), которые не ослепляли клинический диагноз. Консенсус между обоими патологами был достигнут в том случае, если случай был сложным (n = 1). Во всех случаях мы брали образцы лобной, височной, теменной и затылочной коры, миндалины, гиппокампа и поясной извилины, базального ядра, базальных ганглиев, таламуса, среднего мозга, моста, продолговатого мозга и мозжечка.Все пациенты прошли гистологическое исследование гематоксилин-эозином, а также модифицированное окрашивание серебром по Бельшовскому. Иммуногистохимия с антителами к тау (клон AT8 с титром 1: 3750; Endogen, Woburn, Массачусетс), β-амилоид (клон 6F3D с титром 1:20; Novocastra Laboratories, Ньюкасл-апон-Тайн, Англия), α-синуклеин (клон LB509 с титром 1:25; Zymed Laboratories, Inc, Южный Сан-Франциско, Калифорния), убиквитин (поликлональный с титром 1: 100; Dako North America, Inc, Карпинтерия, Калифорния), нейрофиламент (клон 2F11 с титр 1: 800; Dako North America, Inc), TDP-43 (с титром 1: 2000; ProteinTech Group, Inc, Чикаго, Иллинойс) и прионный белок (клон 3F4 с титром 1:50; Dako North America, Inc).Все патологические диагнозы были поставлены на основании ранее опубликованных критериев. 8 -14 Особое внимание уделялось наличию любых других патологических проявлений, включая вторичные нейродегенеративные заболевания (TDP-43-положительные включения, тельца Леви, патологические изменения типа Альцгеймера), амилоидную ангиопатию, сосудистые патологические признаки любых сорт, и аргирофильная болезнь зерна. Все случаи CJD были подтверждены вестерн-блоттингом на наличие прионного белка в Национальном центре наблюдения за патологией прионных заболеваний, Кливленд, Огайо.
Статистический анализ был выполнен с помощью программного обеспечения JMP версии 7.0.0 (SAS Institute Inc, Кэри, Северная Каролина) с установленным α на 0,05. Тест Краскела-Уоллиса использовался для сравнения возраста начала заболевания в различных группах патологической диагностики.
Мы идентифицировали 22 пациента (10 из которых были мужчинами), которые соответствовали нашим критериям включения и исключения (таблица 1). Из этих 22 случаев наиболее частым патологическим диагнозом была CJD (8 случаев [36%]).Мы также обнаружили 5 случаев (23%) с патологическим диагнозом ЛВП с дегенерацией двигательных нейронов (БДН), 4 случая (18%) с таупатией (2 с прогрессирующим надъядерным параличом [PSP] и 2 с кортикобазальной дегенерацией [CBD]). , 3 случая (14%) с DLBD и 2 случая (9%) с AD. Время от начала заболевания до первой неврологической оценки всех 22 случаев составило в среднем 7,5 месяцев.
Гистологические данные представлены в таблице 2. Почти во всех случаях, кроме пациентов с CJD, были вторичные невропатологические признаки, которые включали патологические данные типа Альцгеймера, 10 , 15 сосудистые патологические признаки, амилоидную ангиопатию или болезнь аргирофильного зерна . 16 Однако не было случаев с фокальным глиозом, периваскулярными наручниками или скоплениями микроглии, указывающих на вторичный иммуноопосредованный (паранеопластический) процесс. Тельца Леви отсутствовали в случаях, не связанных с DLBD, и аномальные иммунореактивные поражения TDP-43 не были идентифицированы ни в одном случае, за исключением случаев с FTLD-MND. Во всех случаях FTLD-MND были выявлены иммунореактивные поражения TDP-43 в ядрах XII черепного нерва ствола головного мозга или клетках переднего рога спинного мозга с различными экстрамоторными иммунореактивными включениями TDP-43.
Возраст начала для всех 22 случаев варьировал от 33 до 85 лет и значительно отличался в разных группах ( P = 0,04), причем группа DLBD была самой старой (средний возраст 81 год), а группа FTLD -Группа БПН — самая младшая (средний возраст 50 лет). Оказалось, что существует бимодальное распределение общей продолжительности заболевания: все пациенты с БКЯ умирают через 12 месяцев или раньше после начала, а пациенты с нейродегенеративными заболеваниями, не связанными с БКЯ, имеют продолжительность болезни более 12 месяцев.
Примечательно, что все 3 пациента с DLBD испытали временное послеоперационное или связанное с заболеванием состояние спутанности сознания с очевидным полным выздоровлением, которое примерно на 2 года предшествовало началу деменции. Как только у этих пациентов наступила фаза деменции, смерть наступила быстро, через 4, 11 и 12 месяцев. Таким образом, у этих пациентов было трехфазное течение энцефалопатии, затем бессимптомный или минимально симптоматический двухлетний перерыв и, наконец, фаза быстрого дементирования.Общая продолжительность от начального переходного состояния спутанности сознания до смерти составляла 2,5–3,0 года. Эта временная закономерность была вполне последовательной. Пациент 14 испытал 2 недели острой спутанности сознания после аортокоронарного шунтирования, затем, по-видимому, выздоровел. Через 2 года у него подострое состояние стало слабоумием, и он умер через 4 месяца после начала деменции. У пациента 15 развилось острое состояние спутанности сознания после резекции доброкачественного полипа толстой кишки, и состояние спутанности сознания медленно разрешалось в течение 3 месяцев. Через 2 года у него развилось быстро прогрессирующее слабоумие, и он умер 11 месяцев спустя.Пациент 16 был госпитализирован с кератитом простого герпеса и целлюлитом и пережил недельный эпизод спутанного психоза. Это уменьшилось, но примерно через 2 года у нее развилось быстро прогрессирующее слабоумие, и она умерла через 12 месяцев. Это необычное трехфазное временное течение не наблюдалось ни при каких других нейродегенеративных заболеваниях.
Некоторые клинические признаки, которые часто используются для прогнозирования нейродегенеративного диагноза, не были очень надежными в этой когорте.Паркинсонизм был распространен во всех группах; он присутствовал в 15 из 22 случаев, включая всех 3 пациентов с DLBD и всех 4 пациентов с таупатией. Психоз (зрительные галлюцинации и бред) был зарегистрирован на момент начала деменции у 6 пациентов: 2 с DLBD, 2 с CJD и 2 с AD. Сильные колебания в течение нескольких часов были зарегистрированы у 3 пациентов, 2 с DLBD и 1 с AD. Заболевание двигательных нейронов было зарегистрировано у 3 пациентов, 1 с CJD и 2 с FTLD-MND. Расстройство поведения во сне, связанное с быстрым движением глаз, было зарегистрировано у 3 пациентов: 1 с DLBD и 2 с AD.Миоклонус был специфическим маркером в нашей серии исследований; это было зарегистрировано у 6 пациентов, у всех из которых была CJD. Примечательно, что у 2 пациентов с CJD также были хореоатетоидные движения и апраксия.
Сильные колебания в течение нескольких часов были зарегистрированы у 3 пациентов, 2 с DLBD и 1 с AD. Заболевание двигательных нейронов было зарегистрировано у 3 пациентов, 1 с CJD и 2 с FTLD-MND. Расстройство поведения во сне, связанное с быстрым движением глаз, было зарегистрировано у 3 пациентов: 1 с DLBD и 2 с AD.Миоклонус был специфическим маркером в нашей серии исследований; это было зарегистрировано у 6 пациентов, у всех из которых была CJD. Примечательно, что у 2 пациентов с CJD также были хореоатетоидные движения и апраксия.
Четверо из 14 пациентов с нейродегенеративными заболеваниями, не связанными с CJD, были точно диагностированы при жизни, 2 — с DLBD и 2 — с FTLD-MND. В пятом случае с патологически подтвержденным PSP первоначальный дифференциальный диагноз действительно включал PSP, тогда как окончательный клинический диагноз был CJD. У 6 пациентов окончательный клинический диагноз был в пределах спектра типичных имитаторов, особенно тех, кто относится к спектру расстройств FTLD.Болезнь Крейтцфельда-Якоба считалась первоначальным или окончательным диагнозом у 3 пациентов с нейродегенеративными заболеваниями, не связанными с БКЯ. Напротив, все пациенты с CJD имели правильный диагноз до смерти.
Электроэнцефалография не всегда помогала в установлении правильного диагноза. Периодические атипичные трехфазные волны, резкие волны или эпилептогенные выделения были отмечены в 4 случаях БКЯ. ЭЭГ показала избыток медленных волн в 3 из оставшихся 4 случаев БКЯ, тогда как ЭЭГ была нормальной в 1 случае БКЯ.Все 3 случая DLBD имели атипичные трехфазные волны на ЭЭГ, прерывистые в 2 и полупериодические в другом. При других нейродегенеративных заболеваниях аномалий ЭЭГ не выявлено.
Точно так же результаты CSF имели ограниченную диагностическую ценность. Среди 15 пациентов, прошедших обследование ЦСЖ, повышенные уровни белка были зарегистрированы у 7, в том числе у 1 с DLBD, у 4 с CJD и у 2 с AD; ни у одного пациента не было аномального количества клеток или уровня глюкозы. Уровни нейрон-специфической енолазы были измерены у 14 пациентов и были аномальными (> 35 нг / мл) у 6 из 8 пациентов с CJD (медиана 101.8 нг / мл; диапазон, 39,4-204,0 нг / мл) и 1 пациент с DLBD (35,2 нг / мл). Они были незначительно повышены (20-35 нг / мл) у двух других пациентов с CJD (29,1 нг / мл и 31,7 нг / мл), а также у 2 пациентов с FTLD-MND (23,9 нг / мл и 22,8 нг / мл). ). Уровень нейрон-специфической енолазы был нормальным (<20 нг / мл) у других 3 протестированных пациентов (1 с CBD и 2 с AD). Уровни белка 14-3-3, измеренные с помощью сэндвич-иммунохемилюминометрического анализа, были повышены у 3 из 6 пациентов с CJD и нормальными у 5 пациентов с другими нейродегенеративными заболеваниями, у которых они были проанализированы.
Уровни нейрон-специфической енолазы были измерены у 14 пациентов и были аномальными (> 35 нг / мл) у 6 из 8 пациентов с CJD (медиана 101.8 нг / мл; диапазон, 39,4-204,0 нг / мл) и 1 пациент с DLBD (35,2 нг / мл). Они были незначительно повышены (20-35 нг / мл) у двух других пациентов с CJD (29,1 нг / мл и 31,7 нг / мл), а также у 2 пациентов с FTLD-MND (23,9 нг / мл и 22,8 нг / мл). ). Уровень нейрон-специфической енолазы был нормальным (<20 нг / мл) у других 3 протестированных пациентов (1 с CBD и 2 с AD). Уровни белка 14-3-3, измеренные с помощью сэндвич-иммунохемилюминометрического анализа, были повышены у 3 из 6 пациентов с CJD и нормальными у 5 пациентов с другими нейродегенеративными заболеваниями, у которых они были проанализированы.
Магнитно-резонансная томография головного мозга была в первую очередь диагностической в случаях CJD; диффузионно-взвешенные последовательности магнитного резонанса были аномальными у всех 6 пациентов с CJD, у которых это было выполнено. Это включало диффузно-взвешенные или ослабленные инверсией аномалии восстановления кортикальной гиральной гиперинтенсивности у 5 пациентов и диффузно-взвешенные аномалии в полосатом теле у 4 пациентов, у 2 из которых также были выявлены таламические аномалии. Различные степени атрофии серого вещества были очевидны во всех группах, но имели ограниченную диагностическую ценность.Тяжелая атрофия лобной доли была очевидна у 2 пациентов с ЛВП-БДН, а лобно-теменная атрофия слева больше, чем справа присутствовала у 1 пациента с КБД. Все остальные нейродегенеративные случаи имели генерализованные паттерны атрофии от легкой или легкой до умеренной. Однофотонная эмиссионная компьютерная томография была проведена у 5 пациентов и также имела скромную диагностическую ценность. Среди 3 пациентов с ЛВП-БДН фронтальная гипоперфузия была очевидна у 2, а гипоперфузия левой передней медиальной височной доли наблюдалась у другого.У одного пациента с CBD было выявлено снижение поглощения в лобно-теменных областях, тогда как у другого наблюдалась только лобная гипоперфузия.
Анализ крови у этих пациентов был не очень понятным, хотя у 1 пациента с AD были паранеопластические антинейрональные ядерные антитела 1 типа (ANNA-1 / anti-Hu) и антитела к кальциевым каналам P / Q-типа, с электромиографическими данными, также согласующимися с данными Ламберта-Итона. синдром. Один пациент с PSP имел связывание с рецептором ацетилхолина и антитела к поперечно-полосатым мышцам.Уровни антител к тиропероксидазе были повышены у 4 пациентов, в том числе 2 с CJD, 1 с PSP и 1 с AD. Измерения антимикросомных антител (контрольный диапазон титра <1: 100) были выполнены у 2 пациентов и были повышены у обоих, у одного с CJD (титр 1: 1600), а у другого - с PSP (титр 1: 6400).
Болезнь Крейтцфельдта-Якоба является основным диагностическим критерием среди быстро прогрессирующих нейродегенеративных деменций. Среди наших 22 случаев у пациентов с наиболее агрессивным течением действительно была CJD, и продолжительность болезни была отличительным фактором.Таким образом, все пациенты с деменцией, прогрессирующей до смерти менее чем за 1 год, имели патологические признаки БКЯ, тогда как клиническое течение более 1 года всегда было связано с нейродегенеративным диагнозом, не связанным с БКЯ. Очевидно, что у варианта CJD более длительное течение, но это состояние крайне редко встречается в Соединенных Штатах. 17
Все 3 случая DLBD имели необычное временное течение, однако, это можно было спутать с CJD. Их окончательная фаза дементирования длилась 12 месяцев или меньше.Однако в целом этому предшествовала преходящая энцефалопатия, продолжавшаяся от нескольких недель до нескольких месяцев, за которой следовали примерно 2 года предположительно бессимптомного или минимально симптоматического перерыва. Этот трехфазный курс может быть уникальным для DLBD, поскольку он был задокументирован во всех 3 случаях DLBD и ни в каких других случаях в нашей серии.
Другие исследователи отметили, что нейродегенеративные заболевания составляют менее 5% быстро прогрессирующих деменций. 18 Однако мы подозреваем, что частота будет варьироваться в зависимости от того, как определить quick , который в нашей серии был определен как смерть менее чем через 4 года.
18 Однако мы подозреваем, что частота будет варьироваться в зависимости от того, как определить quick , который в нашей серии был определен как смерть менее чем через 4 года.
Распознаванию CJD помогают другие клинические признаки, которые были очевидны в нашей серии исследований: миоклонус, периодические комплексы на ЭЭГ, восстановление инверсии с ослабленным флюидом и отклонения, взвешенные по диффузии, на магнитно-резонансной томографии. 19 , 20 Хотя повышенный уровень нейрон-специфической энолазы в спинномозговой жидкости 21 также является клиническим ключом к CJD, мы наблюдали повышенный уровень нейрон-специфической энолазы в 1 из наших случаев DLBD, тогда как он был слегка повышен в 2 случаях. Случаи CJD.В нашей серии случаев нейродегенеративных заболеваний, не связанных с CJD, не было аномального уровня белка 14-3-3; следовательно, требуется большая когорта, чтобы определить, может ли белок 14-3-3 быть более специфическим маркером CJD, чем нейрон-специфическая энолаза, когда дифференциальный диагноз включает другие нейродегенеративные заболевания.
Если CJD можно исключить при дифференциальной диагностике быстро прогрессирующей нейродегенеративной деменции, то определенные клинические ключи и результаты тестов могут указывать на конкретный диагноз.Хотя результаты нашей серии исследований требуют повторения, появились наводящие на размышления диагностические данные. Среди наших нейродегенеративных случаев, не связанных с CJD, молодой возраст в начале (около 50 лет) больше всего наводил на мысль о FTLD-MND. И наоборот, наши случаи DLBD характеризовали более пожилой возраст начала заболевания (около 80 лет). Наши пациенты с DLBD также однозначно имели атипичные трехфазные волны на ЭЭГ, которых не было в других нейродегенеративных случаях. В отличие от CJD, аномалии ЭЭГ не имели периодичности, как ранее сообщалось в нескольких случаях DLBD 22 ; однако один из наших случаев DLBD действительно имел полупериодический характер. Таким образом, в течение первого года после начала заболевания аномалии ЭЭГ в контексте быстро прогрессирующей деменции больше указывают на БКЯ, если есть периодические комплексы, тогда как после первого года атипичные трехфазные волны (даже если они полупериодические) могут быть более значительными. указывает на DLBD.
Таким образом, в течение первого года после начала заболевания аномалии ЭЭГ в контексте быстро прогрессирующей деменции больше указывают на БКЯ, если есть периодические комплексы, тогда как после первого года атипичные трехфазные волны (даже если они полупериодические) могут быть более значительными. указывает на DLBD.
Данные о заболевании нижних мотонейронов имеют диагностическое значение, указывая на ЛВП-БДН, если можно исключить БКЯ. Фактически, быстрое течение смерти типично для ЛВП-БДН. 23 В отличие от более типичных ЛВП с длительностью заболевания от 7 до 8 лет, 4 , 24 средняя продолжительность заболевания 2.3 года зарегистрировано в ЛВП-БДН. 23 Таким образом, оценка должна включать тщательный скрининг на заболевание клеток переднего рога и, при необходимости, электромиографию.
Визуализация головного мозга в наших случаях не очень прояснила, за исключением ослабленного жидкостью инверсионного восстановления и отклонений магнитно-резонансной томографии, взвешенных по диффузии, типичных для CJD. Мы не обнаружили какой-либо паттерна атрофии серого вещества, который мог бы служить предиктором быстрого прогрессирования, а паттерны регионарной атрофии не были очень специфичными для точного определения конкретного нейродегенеративного диагноза.В целом, паттерн преимущественно лобной или лобно-височной атрофии более наводит на мысль о БДН-БДН, 25 , тогда как асимметричная лобно-теменная атрофия может немного больше указывать на лежащую в основе КБД. 26 То же самое можно сказать о картинах гипоперфузии на однофотонных эмиссионных компьютерных томографических изображениях.
Почти во всех нейродегенеративных случаях, не связанных с CJD, были очевидны смешанные патологические данные (Таблица 2). Возможно, что эти комбинированные невропатологические находки способствовали быстрому клиническому течению; это включало в основном наложенные патологические данные по болезни Альцгеймера, амилоидную ангиопатию и аргирофильную болезнь зерна. Примечательно, что ранее сообщалось, что амилоидная ангиопатия плюс патологические изменения БА связаны с быстро прогрессирующей деменцией. 27 Патологические признаки болезни Альцгеймера и амилоидная ангиопатия, возможно, также сыграли роль в быстром прогрессировании наших случаев DLBD, поскольку все 3 случая DLBD имели патологию стадии IV Браака 10 и 2 случая имели легкую или умеренную амилоидную ангиопатию. Хотя сообщалось, что и AD, и DLBD иногда имитируют быстрое течение CJD, не было упоминания о наличии вторичных патологических находок в этих случаях. 28
Примечательно, что ранее сообщалось, что амилоидная ангиопатия плюс патологические изменения БА связаны с быстро прогрессирующей деменцией. 27 Патологические признаки болезни Альцгеймера и амилоидная ангиопатия, возможно, также сыграли роль в быстром прогрессировании наших случаев DLBD, поскольку все 3 случая DLBD имели патологию стадии IV Браака 10 и 2 случая имели легкую или умеренную амилоидную ангиопатию. Хотя сообщалось, что и AD, и DLBD иногда имитируют быстрое течение CJD, не было упоминания о наличии вторичных патологических находок в этих случаях. 28
Четыре случая таупатии в нашей серии вызвали наибольшее недоумение. За исключением времени, они представлены не иначе, чем обычно наблюдаемые CBD или PSP, где продолжительность заболевания составляет примерно 7 лет для каждого. 4 Эти случаи также имели вторичные патологические признаки, включая патологические признаки типа Альцгеймера, сосудистые заболевания или аргирофильную болезнь зерна, которые могут играть роль в быстром прогрессировании. Однако стадии БА не были продвинутыми, сосудистые изменения не были серьезными (хотя в 1 случае был микроинфаркт), и в целом аргирофильная болезнь зерна не является маркером агрессивного нейродегенеративного заболевания. 29 Для сравнения этих и более типичных таупатий требуется гораздо большая когорта.
Теоретически, быстрое клиническое течение некоторых из этих нейродегенеративных случаев, не связанных с CJD, могло быть вторично облегчено иммуноопосредованными процессами, направленными на центральную нервную систему. Однако обширное серологическое тестирование, включая паранеопластические антитела, а также спинномозговую жидкость у большинства пациентов, не выявило каких-либо убедительных доказательств этого.У одного пациента действительно были паранеопластические антитела, и, вероятно, это могло внести свой вклад; однако это является исключением. Кроме того, невропатологическое обследование не выявило очагового глиоза, периваскулярных наручников или скоплений микроглии, указывающих на иммуноопосредованный (паранеопластический) процесс.
Это исследование не должно использоваться для определения какой-либо распространенности этих заболеваний, поскольку использование слова «быстрый» могло не охватывать 100% случаев. Кроме того, для проверки некоторых гипотез, выдвинутых нами на основе результатов этого исследования, требуется больший размер выборки.
В заключение, за пределами Соединенного Королевства и Франции, где может быть обнаружен вариант CJD, 17 быстро прогрессирующие нейродегенеративные деменции с выживаемостью более года могут более вероятно представлять собой нейродегенеративное заболевание, не связанное с CJD, хотя пациенты с длительным выживанием со спорадической CJD и заболеванием продолжительность более 12 месяцев. 30 Кроме того, терминальная фаза случаев позднего DLBD может имитировать CJD.
Для корреспонденции: Кейт А.Джозефс, доктор медицины, отделение неврологии, клиника Мэйо, 200 First St SW, Рочестер, Миннесота 55905 ([email protected]).
Принята к публикации: 9 октября 2008 г.
Вклад авторов: Концепция и дизайн исследования : Джозефс, Альског и Крам. Сбор данных : Джозефс, Паризи, Бов, Джаннини и Петерсен. Анализ и интерпретация данных : Джозефс и Альског. Составление рукописи : Джозефс, Альског и Джаннини. Критический пересмотр рукописи на предмет важного интеллектуального содержания : Альског, Паризи, Бов, Крам, Джаннини и Петерсен. Статистический анализ : Джозефс. Получено финансирование : Петерсен. Административная, техническая и материальная поддержка : Parisi and Boeve. Научное руководство : Джаннини.
Раскрытие финансовой информации: Д-р Боев является исследователем в клиническом исследовании, спонсируемом Myriad Pharmaceuticals. Д-р Петерсен был консультантом GE Healthcare и входил в комиссию по мониторингу безопасности данных в клинических испытаниях, спонсируемых Elan Pharmaceuticals.
Финансирование / поддержка: Эта работа была поддержана грантами P50-AG16574 и U01-AG06786 от Национальных институтов здравоохранения и Программой исследования болезни Роберта Х., Кларис Смит и Эбигейл Ван Бурен Фонда Майо. Д-р Джозефс получает поддержку в виде гранта на развитие карьеры в области многодисциплинарных клинических исследований (K12 / NICHD) -HD49078 от Национальных институтов здравоохранения.
Предыдущая презентация: Этот документ был представлен на 133-м ежегодном собрании Американской неврологической ассоциации; 23 сентября 2008 г .; Солт-Лейк-Сити, штат Юта.
1.Американская психиатрическая ассоциация, Диагностическое и статистическое руководство по психическим расстройствам. 4-е изд. Вашингтон, округ Колумбия, Американская психиатрическая ассоциация, 1994;
2.Кнопман DSBoeve Б.Ф. Петерсен RC Основы правильной диагностики легких когнитивных нарушений, деменции и основных подтипов деменции. Mayo Clin Proc 2003; 78 (10) 1290–1308PubMedGoogle ScholarCrossref 3. Баркер WWLuis CAKashuba А и другие.Относительные частоты болезни Альцгеймера, тельца Леви, сосудистой и лобно-височной деменции и склероза гиппокампа в Банке мозга штата Флорида. Alzheimer Dis Assoc Disord 2002; 16 (4) 203–212PubMedGoogle ScholarCrossref 4.Josephs К.А.Петерсен Р.К.Кнопман DS и другие. Клинико-патологический анализ лобно-височной и кортикобазальной дегенераций и ПСП. Неврология 2006; 66 (1) 41-48PubMedGoogle ScholarCrossref 5.Мастера CLHarris ЙОГайдусек ОКРУГ КОЛУМБИЯ и другие. Болезнь Крейтцфельдта-Якоба: закономерности возникновения во всем мире и значение семейной и спорадической кластеризации. Энн Нейрол 1979; 5 (2) 177–188PubMedGoogle ScholarCrossref 6. Ферман
TJSmith
GEBoeve
BF
и другие. Колебания DLB: особенности, которые надежно дифференцируют DLB от БА и нормального старения. Неврология 2004; 62
(2)
181–187PubMedGoogle ScholarCrossref 7.
Ферман
TJSmith
GEBoeve
BF
и другие. Колебания DLB: особенности, которые надежно дифференцируют DLB от БА и нормального старения. Неврология 2004; 62
(2)
181–187PubMedGoogle ScholarCrossref 7.Американская академия медицины сна, Международная классификация нарушений сна. 2-е изд. Вестчестер, штат Иллинойс, Американская академия медицины сна, 2005 год;
8. Мирра С.С.Хейман МакКил D и другие. Консорциум по созданию реестра болезни Альцгеймера (CERAD), часть II: стандартизация невропатологической оценки болезни Альцгеймера. Неврология 1991; 41 (4) 479-486PubMedGoogle ScholarCrossref 9. Хаув JJDaniel SEDickson D и другие.Предварительные нейропатологические критерии NINDS для синдрома Стила-Ричардсона-Ольшевского (прогрессирующий надъядерный паралич). Неврология 1994; 44 (11) 2015–2019PubMedGoogle ScholarCrossref 11.Kretzschmar HAIronside JWDeArmond SJTateishi J Диагностические критерии спорадической болезни Крейтцфельдта-Якоба. Arch Neurol 1996; 53 (9) 913–920PubMedGoogle ScholarCrossref 12. McKeith И.Г. Галаско Д.Косака K и другие.Консенсусные рекомендации по клинической и патологической диагностике деменции с тельцами Леви (DLB): отчет консорциума о международном семинаре DLB. Неврология 1996; 47 (5) 1113–1124PubMedGoogle ScholarCrossref 13. Диксон DWBergeron CChin SS и другие. Управление редких заболеваний Национального института здравоохранения, Управление редких заболеваний, невропатологические критерии кортикобазальной дегенерации. J Neuropathol Exp Neurol 2002; 61 (11) 935–946PubMedGoogle Scholar14. Джозефс
КАПариси
JEKnopman
DS
и другие. Клинически невыявленное заболевание двигательного нейрона при патологически подтвержденной лобно-височной долевой дегенерации с заболеванием двигательного нейрона. Arch Neurol 2006; 63
(4)
506-512PubMedGoogle ScholarCrossref 15.Hyman
BTTrojanowski
Консенсусные рекомендации JQ по посмертной диагностике болезни Альцгеймера от Национального института старения и Рабочей группы Института Рейгана по диагностическим критериям для нейропатологической оценки болезни Альцгеймера. J Neuropathol Exp Neurol 1997; 56
(10)
1095-1097PubMedGoogle ScholarCrossref 16.Braak
HBraak
E Аргирофильные зерна: характерная патология коры головного мозга при старческом слабоумии без изменений Альцгеймера. Neurosci Lett 1987; 76
(1)
124–127PubMedGoogle ScholarCrossref 17. лиры
A Вариант болезни Крейтцфельда-Якоба: риск, неопределенность или безопасность при использовании крови и ее производных? Int Arch Med 2008; 1
(1)
9PubMedGoogle ScholarCrossref 19.Зерр
ISchulz-Schaeffer
WJGiese
А
и другие. Текущий клинический диагноз болезни Крейтцфельдта-Якоба: выявление необычных вариантов. Энн Нейрол 2000; 48
(3)
323- 329PubMedGoogle ScholarCrossref 20. Всемирная организация здравоохранения, Передающиеся губчатые энцефалопатии человека. Wkly Epidemiol Rec 1998; 73
(47)
361- 365PubMedGoogle Scholar21 Санчес-Хуан
PGreen
Аладогана
А
и другие. Тесты спинномозговой жидкости в дифференциальной диагностике болезни Крейтцфельдта-Якоба. Неврология 2006; 67
(4)
637-643PubMedGoogle ScholarCrossref 22.
Джозефс
КАПариси
JEKnopman
DS
и другие. Клинически невыявленное заболевание двигательного нейрона при патологически подтвержденной лобно-височной долевой дегенерации с заболеванием двигательного нейрона. Arch Neurol 2006; 63
(4)
506-512PubMedGoogle ScholarCrossref 15.Hyman
BTTrojanowski
Консенсусные рекомендации JQ по посмертной диагностике болезни Альцгеймера от Национального института старения и Рабочей группы Института Рейгана по диагностическим критериям для нейропатологической оценки болезни Альцгеймера. J Neuropathol Exp Neurol 1997; 56
(10)
1095-1097PubMedGoogle ScholarCrossref 16.Braak
HBraak
E Аргирофильные зерна: характерная патология коры головного мозга при старческом слабоумии без изменений Альцгеймера. Neurosci Lett 1987; 76
(1)
124–127PubMedGoogle ScholarCrossref 17. лиры
A Вариант болезни Крейтцфельда-Якоба: риск, неопределенность или безопасность при использовании крови и ее производных? Int Arch Med 2008; 1
(1)
9PubMedGoogle ScholarCrossref 19.Зерр
ISchulz-Schaeffer
WJGiese
А
и другие. Текущий клинический диагноз болезни Крейтцфельдта-Якоба: выявление необычных вариантов. Энн Нейрол 2000; 48
(3)
323- 329PubMedGoogle ScholarCrossref 20. Всемирная организация здравоохранения, Передающиеся губчатые энцефалопатии человека. Wkly Epidemiol Rec 1998; 73
(47)
361- 365PubMedGoogle Scholar21 Санчес-Хуан
PGreen
Аладогана
А
и другие. Тесты спинномозговой жидкости в дифференциальной диагностике болезни Крейтцфельдта-Якоба. Неврология 2006; 67
(4)
637-643PubMedGoogle ScholarCrossref 22. Haïk
SBrandel
Я.П.Саздович
V
и другие. Деменция с тельцами Леви в группе нейропатологии с подозрением на болезнь Крейтцфельдта-Якоба. Неврология 2000; 55
(9)
1401–1404PubMedGoogle ScholarCrossref 23.Josephs
К.А.Кнопман
DSWhitwell
JL
и другие. Выживаемость при двух вариантах тау-отрицательной лобно-височной долевой дегенерации: FTLD-U против FTLD-MND. Неврология 2005; 65
(4)
645-647PubMedGoogle ScholarCrossref 24.Forman
MSFarmer
Дж. Джонсон
JK
и другие. Лобно-височная деменция: клинико-патологические корреляции. Энн Нейрол 2006; 59
(6)
952- 962PubMedGoogle ScholarCrossref 25.Уитвелл
JLJack
CR
JrSenjem
MLJosephs
KA Паттерны атрофии при патологически подтвержденной ЛВП с дегенерацией мотонейрона и без нее. Неврология 2006; 66
(1)
102-104PubMedGoogle ScholarCrossref 26.Джозефс
KAWhitwell
JLDickson
DW
и другие. Морфометрия на основе вокселей в PSP и CBD, подтвержденных вскрытием. Нейробиол старения 2008; 29
(2)
280–289PubMedGoogle ScholarCrossref 27.Greenberg
SMVonsattel
JPS берет
JW
и другие. Клинический спектр церебральной амилоидной ангиопатии: проявления без крупозного кровоизлияния. Неврология 1993; 43
(10)
2073-2079PubMedGoogle ScholarCrossref 28.Tschampa
HJNeumann
МЗерр
я
и другие.Пациенты с болезнью Альцгеймера и деменцией с тельцами Леви, ошибочно принимаемыми за болезнь Крейтцфельда-Якоба. J Neurol Neurosurg Psychiatry 2001; 71
(1)
33–39PubMedGoogle ScholarCrossref 29.
Haïk
SBrandel
Я.П.Саздович
V
и другие. Деменция с тельцами Леви в группе нейропатологии с подозрением на болезнь Крейтцфельдта-Якоба. Неврология 2000; 55
(9)
1401–1404PubMedGoogle ScholarCrossref 23.Josephs
К.А.Кнопман
DSWhitwell
JL
и другие. Выживаемость при двух вариантах тау-отрицательной лобно-височной долевой дегенерации: FTLD-U против FTLD-MND. Неврология 2005; 65
(4)
645-647PubMedGoogle ScholarCrossref 24.Forman
MSFarmer
Дж. Джонсон
JK
и другие. Лобно-височная деменция: клинико-патологические корреляции. Энн Нейрол 2006; 59
(6)
952- 962PubMedGoogle ScholarCrossref 25.Уитвелл
JLJack
CR
JrSenjem
MLJosephs
KA Паттерны атрофии при патологически подтвержденной ЛВП с дегенерацией мотонейрона и без нее. Неврология 2006; 66
(1)
102-104PubMedGoogle ScholarCrossref 26.Джозефс
KAWhitwell
JLDickson
DW
и другие. Морфометрия на основе вокселей в PSP и CBD, подтвержденных вскрытием. Нейробиол старения 2008; 29
(2)
280–289PubMedGoogle ScholarCrossref 27.Greenberg
SMVonsattel
JPS берет
JW
и другие. Клинический спектр церебральной амилоидной ангиопатии: проявления без крупозного кровоизлияния. Неврология 1993; 43
(10)
2073-2079PubMedGoogle ScholarCrossref 28.Tschampa
HJNeumann
МЗерр
я
и другие.Пациенты с болезнью Альцгеймера и деменцией с тельцами Леви, ошибочно принимаемыми за болезнь Крейтцфельда-Якоба. J Neurol Neurosurg Psychiatry 2001; 71
(1)
33–39PubMedGoogle ScholarCrossref 29. Togo
Тисодзима
DAkatsu
ЧАС
и другие. Клинические особенности аргирофильной болезни зерна: ретроспективный обзор случаев с психоневрологическими симптомами. Am J Geriatr Psychiatry 2005; 13
(12)
1083-1091PubMedGoogle ScholarCrossref 30.Parchi
PGiese
ACapellari
S
и другие.Классификация спорадической болезни Крейтцфельдта-Якоба на основе молекулярного и фенотипического анализа 300 субъектов. Энн Нейрол 1999; 46
(2)
224–233PubMedGoogle ScholarCrossref
Togo
Тисодзима
DAkatsu
ЧАС
и другие. Клинические особенности аргирофильной болезни зерна: ретроспективный обзор случаев с психоневрологическими симптомами. Am J Geriatr Psychiatry 2005; 13
(12)
1083-1091PubMedGoogle ScholarCrossref 30.Parchi
PGiese
ACapellari
S
и другие.Классификация спорадической болезни Крейтцфельдта-Якоба на основе молекулярного и фенотипического анализа 300 субъектов. Энн Нейрол 1999; 46
(2)
224–233PubMedGoogle ScholarCrossrefБыстро прогрессирующая деменция | Hometouch
Деменция — это прогрессирующее заболевание, которое со временем ухудшается. Скорость ухудшения зависит от человека. Возраст, общее состояние здоровья и основное заболевание, вызывающее повреждение мозга, — все это влияет на характер прогрессирования.Однако для некоторых людей спад может быть внезапным и быстрым.
Ухудшение деменции обычно медленное и постепенное. Быстро прогрессирующие деменции или РПЗ бывают разными. Они быстро прогрессируют, часто в течение недель или месяцев, и могут быстро оказаться фатальными. Быстро прогрессирующие деменции редки, но они могут быть очень неприятными для человека, страдающего этим заболеванием, и для тех, кто любит их и заботится о них.
Почему деменция может быстро прогрессировать?
Болезнь Альцгеймера обычно прогрессирует медленно и постепенно, в то время как у людей, страдающих сосудистой деменцией, наблюдается периодическое постепенное нарушение функций.Однако многие факторы влияют на развитие деменции. Генетическое наследие человека будет иметь значение, так же как и его общее физическое здоровье. Люди с сердечно-сосудистыми заболеваниями или диабетом, особенно если они плохо контролируются, подвержены риску более быстрого ухудшения состояния. Также уязвимы люди с ослабленным иммунитетом и рецидивирующими инфекциями. Деменция с ранним началом имеет тенденцию прогрессировать быстрее. Люди, у которых слабоумие развивается в возрасте от тридцати до пятидесяти лет, по всей видимости, живут на два года меньше, чем те, у которых деменция диагностируется в более позднем возрасте.
Большинство случаев внезапной дезориентации и быстро прогрессирующей деменции у пожилых людей вызваны инфекционным делирием. Инфекции мочевыводящих путей и пневмония могут спровоцировать острую спутанность сознания, которая быстро наступает, в результате чего люди становятся бессвязными, запутанными и дезориентированными. Также распространены возбуждение, агрессия и странное поведение. Хорошая новость заключается в том, что симптомы делирия можно обратить вспять, если лечить инфекцию соответствующим образом.
Может ли это быть быстро прогрессирующее слабоумие?
RPD бывает сложно диагностировать.Однако точная диагностика этих состояний имеет решающее значение для выявления любых излечимых причин и защиты от дальнейшего повреждения клеток головного мозга. Раннее обследование в больнице, проведенное специалистом, может помочь выявить проблемы и начать соответствующее лечение, если это возможно. Рак, инфекции, токсины и аутоиммунные состояния могут вызывать быстрое снижение умственной функции, а также более распространенные нейродегенеративные причины деменции, такие как болезнь Альцгеймера, инсульты и болезнь Паркинсона.
Исследование RPD
При оценке RPD врач подробно изучает историю болезни, чтобы узнать о симптомах и проблемах, с которыми сталкивается человек. Информация об истории болезни, здоровье близких родственников и недавнем воздействии химикатов, инфекций или лекарств может помочь выявить любые потенциальные причины. Тщательное обследование может помочь врачу выявить любые физические проблемы и оценить текущий уровень психических функций.
Исследования обычно включают лабораторные анализы крови, мочи и жидкости, окружающей головной и спинной мозг.Мозг будет сканироваться с использованием устройства МРТ или КТ, и может быть проведена электроэнцефалограмма или ЭЭГ для измерения электрической активности в головном мозге. Вся эта информация будет усвоена и рассмотрена, чтобы помочь диагностировать причину деменции.
Вся эта информация будет усвоена и рассмотрена, чтобы помочь диагностировать причину деменции.
Что происходит при быстро прогрессирующей деменции?
Представление и прогресс RPD будут различаться у разных людей. У пострадавших обычно возникают проблемы с памятью, мыслительными процессами и общением. У многих людей также есть изменения личности или поведения, а также расстройство настроения.Изменения движений также могут возникать в результате повреждения клеток головного мозга.
Некоторые формы РПЗ поддаются лечению, и при быстрой постановке диагноза ранние симптомы могут быть обращены вспять. К сожалению, для других причин этого состояния нет доступных лекарств. Неизбежно нарастание симптомов и снижение функций. К сожалению, в течение нескольких месяцев или лет быстро прогрессирующая деменция приведет к отказу всех систем организма и смерти.
Что вызывает RPD?
Существует множество причин РПЗ, в том числе:
- Аутоиммунные заболевания , при которых иммунная система организма активируется для борьбы с собственными клетками.При некоторых аутоиммунных заболеваниях вырабатываются антитела, которые могут атаковать мозг или нервную ткань.
- Необычные проявления общих причин деменции. В редких случаях болезнь Альцгеймера, Паркинсона, лобно-височная деменция и деменция с тельцами Леви могут развиваться быстрее и прогрессировать быстрее.
- Гормональные нарушения и нарушения обмена веществ.
- Прионные болезни. Это редкие нейродегенеративные заболевания, вызванные аномально сложенными белками в головном мозге.Прионные заболевания включают болезнь Крейтцфельдта-Якоба (CJD) и фатальную семейную бессонницу. Механизмы заражения плохо изучены.
- Инфекции.
- Нарушение кровотока в головном мозге , включая инсульты, вызывающие более внезапную и тяжелую сосудистую деменцию.
- Структурные проблемы головного мозга , например гидроцефалия.
- Токсины.
- Дефицит витаминов.
- Рак.
- Побочные эффекты или осложнения от назначенных лекарств.
- Рецидивирующие припадки или припадки.

Что такое лечение РПЗ?
Правильное лечение RPD будет зависеть от типа деменции и причины состояния. Для RPD, вызванной раком, гормональным дисбалансом или метаболическими нарушениями, лечение, направленное на первопричину, также может помочь в лечении деменции. К сожалению, для многих людей нет лекарства, однако лечение может помочь уменьшить стресс, сохранить комфорт и улучшить качество жизни.>
Хотите узнать больше о высококачественной услуге домашнего ухода
Hometouch ? Найдите помощника по уходу Запишитесь на звонокОценка быстро прогрессирующей деменции, достигающей кульминации при диагностике болезни Крейтцфельдта – Якоба
Быстро прогрессирующая деменция — любопытное и неуловимое клиническое описание модели когнитивных нарушений, которые прогрессируют быстрее, чем типичные синдромы деменции. Дифференциальный диагноз и клиническое обследование быстро прогрессирующей деменции довольно обширны и включают поиск инфекционных, воспалительных, аутоиммунных, неопластических, метаболических и нейродегенеративных причин.Мы представляем случай 76-летнего человека, ранее имевшего высокую функциональность, у которого в течение 6 месяцев была быстро прогрессирующая деменция. Его наиболее заметными симптомами были когнитивные нарушения, афазия, зрительные галлюцинации и атаксия. После обширного набора тестов в больнице дифференциальный диагноз оставался вероятным: CJD по сравнению с аутоиммунным энцефалитом. Он клинически ухудшился и прогрессировал до акинетического мутизма и миоклонуса. Он скончался через 8 недель после того, как впервые попал в больницу, и вскрытие подтвердило диагноз спорадической CJD.Мы используем этот иллюстративный случай в качестве основы для обсуждения клинических и диагностических соображений при обследовании быстро прогрессирующей деменции. Мы также более подробно обсуждаем CJD и аутоиммунный энцефалит, две основные диагностические возможности у нашего пациента.
Мы также более подробно обсуждаем CJD и аутоиммунный энцефалит, две основные диагностические возможности у нашего пациента.
1. Случай
76-летний профессор лингвистики, мужчина, был направлен в службу неотложной терапии в связи с ухудшением состояния спутанности сознания в течение 2 недель. Он был независимым в своей повседневной деятельности (ADL) и своей инструментальной повседневной деятельности (IADL) на исходном уровне 6 месяцев назад.Семья одобрила снижение когнитивных способностей, которое началось с проблем с памятью, трудностей с поиском слов и неустойчивой походки. Они также подтвердили, что в анамнезе были ажитация и галлюцинации по ночам. За 2 недели до обращения в отделение неотложной помощи его симптомы прогрессировали еще более быстрыми темпами, он был слишком слаб, чтобы ходить, и у него возникло новое недержание мочи и стула. До тех пор, пока его когнитивный дефицит не ухудшился, он все еще работал профессором лингвистики на уровне послесреднего образования.
В его анамнезе была ишемическая болезнь сердца, гипертония, диабет 2 типа, астма и доброкачественная гиперплазия простаты. В личном или семейном анамнезе злокачественных новообразований или деменции не было. Он никогда не проходил обследование на злокачественные новообразования. Не было истории охоты или употребления в пищу мяса дичи. Его лекарства включали АСК, кандесартан, гидрохлоротиазид, метформин, глимепирид, препараты железа, поливитамины и глазные капли тимолол. Не было никаких безрецептурных лекарств, запрещенных наркотиков или алкоголя.На экзамене его жизненно важные органы были стабильными. Его слизистые оболочки были сухими, а JVP был плоским. Его кардиологические, респираторные и абдоминальные обследования не были примечательными. Его неврологический осмотр выявил небольшой паралич взгляда вверх и гипертонус, зависящий от скорости движения, в верхних конечностях. Фасцикуляций и миоклонусов не было. Рефлексы и ощущения не нарушены.
Его количество лейкоцитов было 2,7 × 10 9 (нормальное 3,5–10,5), его гемоглобин был 134 г / л, а его тромбоциты были 196 × 10 9 . Электролиты и расширенные электролиты были в пределах нормы, за исключением натрия 125 ммоль / л (норма 136–145). LTF и билирубин были в пределах нормы, ТТГ — 2,35 (норма), а уровень B12 — 278 пмоль / л (норма). Серологии на сифилис и ВИЧ были отрицательными, как и антинуклеарные антитела (ANA). МРТ с диффузионным взвешиванием показала диффузную потерю объема паренхимы, которая была заметной для возраста и легких микроангиопатических изменений. Его ЭЭГ была ненормальной, но неспецифической с нерегулярными периодами тета-активности 6-7 Гц, смешанными с короткими дельта-ритмами 2-4 Гц, наиболее заметными в лобных областях.Не было альфа-активности или очевидных эпилептиформных, фокальных или латерализующих особенностей. В спинномозговой жидкости было обнаружено 6 ядерных клеток (норма 0–5), нормальная глюкоза и немного повышенный уровень белка 0,55 г / л (нормальный 0,15–0,45 г / л). Олигоклональные полосы в спинномозговой жидкости не обнаружены. ЦСЖ была отрицательной по тау и белку 14-3-3, но положительной по конечной точке преобразования, вызванной сотрясением (EP-QuIC) в Национальной микробиологической лаборатории в Виннипеге. Панель паранеопластических антител (анти-hu, ri, yo, ma2, cv2 и амфифизин) (митоген) была отрицательной.
Электролиты и расширенные электролиты были в пределах нормы, за исключением натрия 125 ммоль / л (норма 136–145). LTF и билирубин были в пределах нормы, ТТГ — 2,35 (норма), а уровень B12 — 278 пмоль / л (норма). Серологии на сифилис и ВИЧ были отрицательными, как и антинуклеарные антитела (ANA). МРТ с диффузионным взвешиванием показала диффузную потерю объема паренхимы, которая была заметной для возраста и легких микроангиопатических изменений. Его ЭЭГ была ненормальной, но неспецифической с нерегулярными периодами тета-активности 6-7 Гц, смешанными с короткими дельта-ритмами 2-4 Гц, наиболее заметными в лобных областях.Не было альфа-активности или очевидных эпилептиформных, фокальных или латерализующих особенностей. В спинномозговой жидкости было обнаружено 6 ядерных клеток (норма 0–5), нормальная глюкоза и немного повышенный уровень белка 0,55 г / л (нормальный 0,15–0,45 г / л). Олигоклональные полосы в спинномозговой жидкости не обнаружены. ЦСЖ была отрицательной по тау и белку 14-3-3, но положительной по конечной точке преобразования, вызванной сотрясением (EP-QuIC) в Национальной микробиологической лаборатории в Виннипеге. Панель паранеопластических антител (анти-hu, ri, yo, ma2, cv2 и амфифизин) (митоген) была отрицательной.
У него развились миоклонус и мутизм, и он был выписан в паллиативную лечебницу. Он скончался через 8 недель после обращения в отделение неотложной помощи. Посмертное вскрытие головного мозга показало микроспонгиоз, потерю нейронов и глиоз в коре, гиппокампе, базальных ганглиях и мозжечке, что соответствует sCJD.
2. Быстро прогрессирующая деменция
В настоящее время не существует общепринятого определения случая быстро прогрессирующей деменции.Некоторые авторы предполагают, что деменция, которая проявляется и прогрессирует в течение 2 лет, должна рассматриваться как быстро прогрессирующая [1], в то время как другие утверждают, что когнитивные нарушения, протекающие быстрее, чем типичная болезнь Альцгеймера или сосудистая деменция, должны вызывать подозрение на быстрое развитие болезни.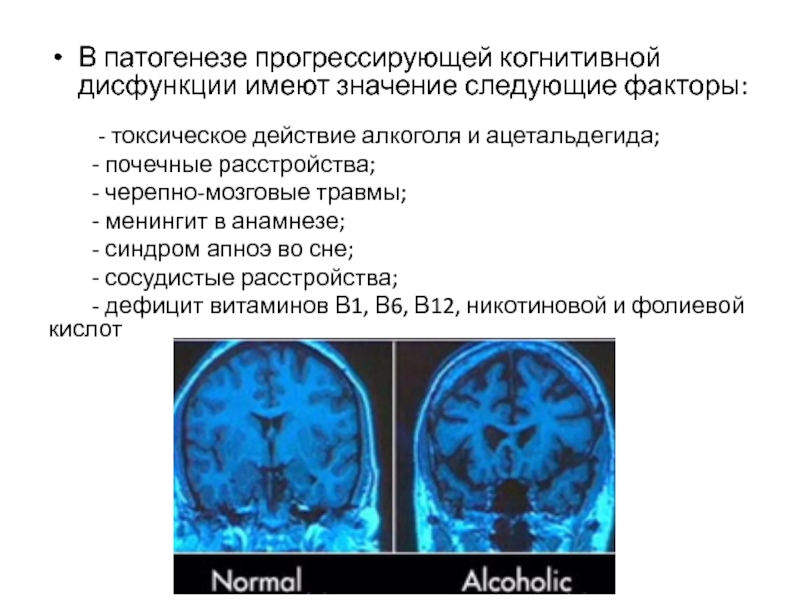 синдром деменции [2–4]. Дифференциальный диагноз быстро прогрессирующей деменции довольно обширен и включает в себя инфекционные, воспалительные, аутоиммунные, неопластические, метаболические и нейродегенеративные заболевания.
синдром деменции [2–4]. Дифференциальный диагноз быстро прогрессирующей деменции довольно обширен и включает в себя инфекционные, воспалительные, аутоиммунные, неопластические, метаболические и нейродегенеративные заболевания.
Клиническая оценка предполагаемого быстро прогрессирующего синдрома деменции должна начинаться с тщательного сбора анамнеза пациента с упором на выяснение первых неврологических симптомов и установление точного временного курса, включая новые дефициты [5–7].
Клиницисты должны также узнать о лекарствах, особенно о холинолитиках и бензодиазепинах, а также о незаконном употреблении наркотиков и алкоголя [6]. Крайне важно получить сопутствующий анамнез от друзей и семьи, а также обзор систем с акцентом на другие пораженные системы органов [6].Медицинский осмотр должен быть направлен на выявление вегетативной дисфункции, экстрапирамидных признаков, фасцикуляций и миоклонуса, а также на выявление стигматов метаболических и неопластических заболеваний [1, 6].
Существует множество диагностических тестов, которые можно включить в исследование быстро прогрессирующего синдрома деменции. Выбор и выбор времени дополнительных тестов должны производиться разумно и поэтапно. Делирий, инфекционные и метаболические энцефалопатии должны стать объектами первоначальных исследований [2, 3].Следующие уровни тестирования должны быть направлены на поиск аутоиммунной и нейродегенеративной этиологии [2, 3]. Наконец, тестирование может быть расширено для поиска редких и необычных проявлений заболеваний, включая атипичные инфекции, в зависимости от элементов истории болезни и воздействия, а также аномальных результатов предыдущих этапов исследования [2, 3].
Исследования следует начинать с рутинных лабораторных исследований и визуализационных исследований, направленных на выявление общих, обратимых состояний [5, 8].Общий анализ крови, электролиты, повышенный уровень электролитов, B12, ТТГ, общий анализ мочи, посевы крови и мочи, рентген грудной клетки и компьютерная томография должны быть заказаны заранее при обследовании пациента с возможным быстро прогрессирующим слабоумием, чтобы помочь отличить деменцию. от бреда [5]. Следует выполнить люмбальную пункцию и отправить СМЖ на подсчет клеток, бактериологию и биохимический анализ для оценки менингита [1, 5]. ЦСЖ также следует направлять для получения 14-3-3, тау-белков и EP-QuIC (конечный тест сотрясения) для оценки CJD / прионной болезни [1, 3, 5].МРТ головного мозга с последовательностью FLAIR полезен для оценки аутоиммунных, нейродегенеративных и неопластических причин быстро прогрессирующей деменции [1, 3]. Панели паранеопластических и аутоиммунных антител также должны быть протестированы [1]. Электроэнцефалограмма (ЭЭГ) полезна для оценки прионов и нейродегенеративных заболеваний [1, 3, 5].
от бреда [5]. Следует выполнить люмбальную пункцию и отправить СМЖ на подсчет клеток, бактериологию и биохимический анализ для оценки менингита [1, 5]. ЦСЖ также следует направлять для получения 14-3-3, тау-белков и EP-QuIC (конечный тест сотрясения) для оценки CJD / прионной болезни [1, 3, 5].МРТ головного мозга с последовательностью FLAIR полезен для оценки аутоиммунных, нейродегенеративных и неопластических причин быстро прогрессирующей деменции [1, 3]. Панели паранеопластических и аутоиммунных антител также должны быть протестированы [1]. Электроэнцефалограмма (ЭЭГ) полезна для оценки прионов и нейродегенеративных заболеваний [1, 3, 5].
3. Болезнь Крейтцфельдта – Якоба
Прионные болезни человека встречаются довольно редко: во всем мире заболеваемость составляет 0,5–1 случай на миллион человек [9, 10]. Существуют генетические, приобретенные и спорадические подтипы прионной болезни [4, 9, 11, 12].Генетическими формами прионной болезни являются фатальная семейная бессонница и болезнь Герстмана – Штрауслера – Шейнкера [4, 9, 11, 12]. Приобретенные прионные заболевания включают куру, ятрогенный и вариантный БКЯ [4, 9, 13]. Спорадическая CJD является наиболее распространенной и составляет 85–90% всех прионных заболеваний человека [10, 13, 14].
CJD может проявляться быстрым снижением когнитивных функций, нарушением походки, зрительными и поведенческими расстройствами и может прогрессировать до миоклонуса и акинетического мутизма [8, 10, 11, 13, 14]. CJD обычно присутствует в 6 -м или 7 -м десятилетиях жизни; случаи, представленные в возрасте до 30 или после 80 лет, чрезвычайно редки [14, 15].CJD одинаково поражает мужчин и женщин [10, 14]. CJD имеет быстро прогрессирующее течение и всегда приводит к летальному исходу [15]. Средняя выживаемость при sCJD составляет 5 месяцев, при этом 90% пациентов умирают в течение 1 года [1, 4, 10, 14–16].
Поскольку CJD относительно редко и незнакома большинству клиницистов, диагноз затруднен, и CJD часто ошибочно диагностируется.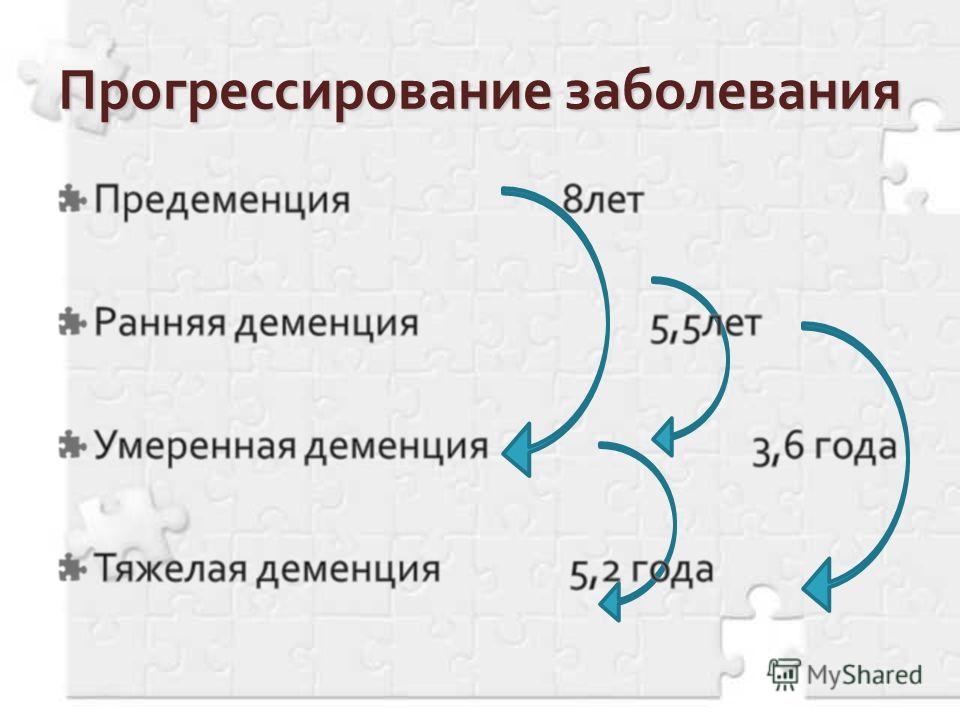 CJD вызывается прионами, которые представляют собой псевдоинфекционные, самораспространяющиеся белковые частицы, вызывающие агрегацию, губчатые изменения и потерю нейронов [11, 12, 14, 15].Окончательный диагноз CJD ставится на основании гистопатологического анализа ткани головного мозга, полученной при биопсии головного мозга или, чаще, при аутопсии [1, 7, 10, 12, 15]. Поддерживающие методы диагностики, которые могут подтвердить диагноз CJD, включают ЭЭГ, МРТ, CSF 14-3-3 и EP-QuIC. Ранняя CJD может проявляться как неспецифическое замедление на ЭЭГ, тогда как характерные периодические трифашические резкие волновые комплексы могут проявляться позже в течении болезни [3, 4, 11, 12, 15]. Чувствительность и специфичность ЭЭГ для выявления БКЯ составляет 50–66% и 74–91% соответственно [3, 12].Признак пульвинара на МРТ относится к двусторонней гиперинтенсивности FLAIR в ядрах пульвинара и таламуса и может быть замечен при вариантной и спорадической БКЯ [7, 9, 10, 12]. МРТ имеет 91% чувствительность и 95% специфичность для CJD [3, 12, 15]. 14-3-3 и тау-белки в спинномозговой жидкости чувствительны к повреждению нейронов, хотя они не специфичны для CJD [7, 12]. EP-QuIC — это эмпирически подтвержденный анализ, который использует внутренние свойства связанного с заболеванием прионного белка в спинномозговой жидкости пациентов, чтобы вызвать неправильную укладку и агрегацию рекомбинантного прионного белка.Белковые агрегаты взаимодействуют с красителем, что приводит к заметным изменениям его флуоресценции [17]. EP-QuIC имеет 80–90% чувствительность и 99–100% специфичность для диагностики прионной болезни [4, 12].
CJD вызывается прионами, которые представляют собой псевдоинфекционные, самораспространяющиеся белковые частицы, вызывающие агрегацию, губчатые изменения и потерю нейронов [11, 12, 14, 15].Окончательный диагноз CJD ставится на основании гистопатологического анализа ткани головного мозга, полученной при биопсии головного мозга или, чаще, при аутопсии [1, 7, 10, 12, 15]. Поддерживающие методы диагностики, которые могут подтвердить диагноз CJD, включают ЭЭГ, МРТ, CSF 14-3-3 и EP-QuIC. Ранняя CJD может проявляться как неспецифическое замедление на ЭЭГ, тогда как характерные периодические трифашические резкие волновые комплексы могут проявляться позже в течении болезни [3, 4, 11, 12, 15]. Чувствительность и специфичность ЭЭГ для выявления БКЯ составляет 50–66% и 74–91% соответственно [3, 12].Признак пульвинара на МРТ относится к двусторонней гиперинтенсивности FLAIR в ядрах пульвинара и таламуса и может быть замечен при вариантной и спорадической БКЯ [7, 9, 10, 12]. МРТ имеет 91% чувствительность и 95% специфичность для CJD [3, 12, 15]. 14-3-3 и тау-белки в спинномозговой жидкости чувствительны к повреждению нейронов, хотя они не специфичны для CJD [7, 12]. EP-QuIC — это эмпирически подтвержденный анализ, который использует внутренние свойства связанного с заболеванием прионного белка в спинномозговой жидкости пациентов, чтобы вызвать неправильную укладку и агрегацию рекомбинантного прионного белка.Белковые агрегаты взаимодействуют с красителем, что приводит к заметным изменениям его флуоресценции [17]. EP-QuIC имеет 80–90% чувствительность и 99–100% специфичность для диагностики прионной болезни [4, 12].
Диагноз вероятной CJD требует быстро прогрессирующего слабоумия и двух из четырех из четырех: миоклонии, визуальных или мозжечковых симптомов, пирамидных / экстрапирамидных симптомов или акинетического мутизма и положительного результата вспомогательного теста (ЭЭГ, 14-3-3 или МРТ) [14, 15]. EP-QuIC — это новый диагностический тест, который еще не включен в диагностические критерии ВОЗ для CJD [14, 15].
Все подтипы CJD прогрессируют и однозначно приводят к смерти. Помимо лечения, направленного на контролирование симптомов, не существует эффективных методов лечения, которые останавливают или препятствуют прогрессированию заболевания [7]. Несмотря на ограниченные возможности лечения, установление диагноза прионной болезни имеет большое значение. В большинстве юрисдикций CJD — это заболевание, подлежащее регистрации, которое исследуется органами общественного здравоохранения [14]. Диагностика семейных вариантов заболевания имеет значение для генетического тестирования членов семьи.Что касается оказания непосредственной помощи пациенту, после постановки диагноза CJD выявляется четкая траектория заболевания. Пациентов и их семьи следует направлять к врачам, прошедшим подготовку по оказанию паллиативной помощи, для получения помощи при переходе на хосписную помощь [14, 18].
4. Аутоиммунный энцефалит
Аутоиммунный энцефалит — важная этиология заболевания, которая включается в дифференциальный диагноз быстро прогрессирующей деменции. Первоначальные проявления аутоиммунного энцефалита сильно различаются и зависят от антител, вызывающих заболевание, и ассоциированного заболевания.Психиатрические проявления и снижение когнитивных функций являются наиболее часто наблюдаемыми начальными симптомами аутоиммунного энцефалита [3, 19]. Существует несколько подгрупп аутоиммунного энцефалита: классический паранеопластический энцефалит, заболевания, связанные с аутоантителами против ионных каналов, заболевания, связанные с аутоантителами против внутриклеточных синаптических белков, и, наконец, аутоиммунный энцефалит, при котором антигены четко не определены [19].
Аутоиммунные антитела против внеклеточных эпитопов ионных каналов по своей природе являются патологическими; Помимо снижения когнитивных функций, они также могут приводить к уникальным клиническим проявлениям [3, 19].Лимбический энцефалит с богатым антилейцином глиомой 1 (LGI1) вызывается антителами против элемента LGI1 комплекса потенциал-управляемых калиевых каналов (VGKC) [20]. Энцефалит LGI1 проявляется судорогами, дизавтономией и гипонатриемией, вторичными по отношению к SIADH [3, 6, 20, 21]. Явный психоз и вегетативная дисфункция являются особенностями энцефалита рецепторов N-метил-D-аспартата (NMDA); это связано с тератомой яичника и раком яичек [3].
Энцефалит LGI1 проявляется судорогами, дизавтономией и гипонатриемией, вторичными по отношению к SIADH [3, 6, 20, 21]. Явный психоз и вегетативная дисфункция являются особенностями энцефалита рецепторов N-метил-D-аспартата (NMDA); это связано с тератомой яичника и раком яичек [3].
В отличие от аутоиммунных антител против ионных каналов, паранеопластические антитела сами по себе не являются патологическими.Психоневрологические симптомы, включая тревогу и галлюцинации с колеблющимся течением, как правило, присутствуют при паранеопластическом энцефалите [1, 3, 19]. Паранеопластические антитела часто предшествуют диагностике основного злокачественного новообразования, иногда на 1 год и более [2, 19].
GAD65 (декарбоксилаза глутаминовой кислоты) аутоиммунный энцефалит является примером заболевания, связанного с аутоантителами против внутриклеточного синаптического белка [3, 19]. Энцефалит GAD65 проявляется синдромом скованности, прогрессирующей ригидностью и миоклонусом туловищных мышц [3, 19].Интересно, что болезнь GAD65 также связана с резистентной к лечению эпилепсией и новым началом диабета 1 типа [3, 19].
В отличие от CJD, который обычно имеет мягкую биохимию CSF, CSF при аутоиммунном энцефалите обычно демонстрирует лимфоцитарный плеоцитоз с повышенным содержанием белка и случайным присутствием олигоклональных полос [3]. ЭЭГ и МРТ гораздо менее полезны для постановки диагноза аутоиммунного энцефалита по сравнению с CJD. МРТ часто бывает нормальным в случаях аутоиммунного энцефалита и не может исключить этот диагноз [19].ЭЭГ полезна для мониторинга судорожной активности, связанной с некоторыми формами аутоиммунного энцефалита; специфические данные ЭЭГ могут наблюдаться при энцефалите NMDAR и лимбическом энцефалите LGI1 (инактивированная лейцином глиома) [19]. Гистопатология не является ни практичной, ни специфической для аутоиммунного энцефалита [19].
Аутоиммунный энцефалит остается важным диагностическим аспектом при оценке быстро прогрессирующей деменции, поскольку он поддается лечению и потенциально обратим. Большинство аутоиммунных энцефалитов, как правило, поддаются лечению стероидами, обычно назначаемыми в виде солюмедрола 1 г внутривенно в течение 3-5 дней с последующим постепенным снижением дозы [1, 3, 6].Также можно рассмотреть возможность плазмообменной терапии и внутривенного иммуноглобулина (ВВИГ), в первую очередь при подтвержденном или предполагаемом аутоиммунном энцефалите, который не отвечает только на стероиды [1, 3, 6, 19]. Терапия второй линии для лечения аутоиммунного энцефалита включает ритуксимаб и циклофосфамид [3, 19]. Следует отметить, что паранеопластический энцефалит обычно не поддается лечению стероидами или сплетением, но может улучшиться при лечении ассоциированного злокачественного новообразования [3, 19].
Большинство аутоиммунных энцефалитов, как правило, поддаются лечению стероидами, обычно назначаемыми в виде солюмедрола 1 г внутривенно в течение 3-5 дней с последующим постепенным снижением дозы [1, 3, 6].Также можно рассмотреть возможность плазмообменной терапии и внутривенного иммуноглобулина (ВВИГ), в первую очередь при подтвержденном или предполагаемом аутоиммунном энцефалите, который не отвечает только на стероиды [1, 3, 6, 19]. Терапия второй линии для лечения аутоиммунного энцефалита включает ритуксимаб и циклофосфамид [3, 19]. Следует отметить, что паранеопластический энцефалит обычно не поддается лечению стероидами или сплетением, но может улучшиться при лечении ассоциированного злокачественного новообразования [3, 19].
5. Заключение
Быстро прогрессирующая деменция — интересный клинический сценарий с множеством диагностических возможностей.Это требует тщательного клинического обследования и диагностического обследования, которое должно быть сосредоточено на обнаружении обратимых излечимых состояний. CJD и аутоиммунный энцефалит включены в дифференциальный диагноз быстро прогрессирующей деменции. Эти два заболевания могут проявляться примерно одинаково; однако они резко различаются по своей клинической траектории и прогнозу. Аутоиммунный энцефалит проявляется нейропсихиатрическими симптомами и хорошо поддается лечению стероидами, тогда как CJD проявляется снижением когнитивных функций, экстрапирамидными симптомами и нарушением походки, а также миоклонусом; он всегда приводит к летальному исходу, обычно в течение 12 месяцев после появления симптомов.
Конфликт интересов
Авторы заявляют об отсутствии конфликта интересов.
Вклад авторов
Пармвир Пармар и Даниэль Кобевка оказывали помощь пациентам и писали, рецензировали и готовили рукопись для подачи вместе с Кертисом Л. Купером.
Быстро прогрессирующая деменция и атаксия у пожилого мужчины
Изучение дела (доктор Р. М. Ахмед)
История
76-летний мужчина, инженер на пенсии с университетским образованием, был направлен с двухмесячной историей ухудшения баланса и памяти. Принимал метформин при сахарном диабете 2 типа. Он пришел с женой в течение 2 дней после направления и принес свои недавние снимки МРТ головного мозга и брюшной полости. Никакие проблемы с памятью его не беспокоили (его жена, конечно, была), но при ходьбе чувствовал себя неуравновешенным. Стало очевидно, что он фактически терял равновесие и память в течение примерно 12 месяцев, но за последние 2 месяца это прогрессировало быстрее. У него не было ни головных болей, ни сенсорных симптомов, ни симптомов мочеиспускания. Он хорошо спал и ел, не похудел.Он был опытным пианистом, и его музыкальные способности не ухудшились. Он никогда не курил и мало пил.
Принимал метформин при сахарном диабете 2 типа. Он пришел с женой в течение 2 дней после направления и принес свои недавние снимки МРТ головного мозга и брюшной полости. Никакие проблемы с памятью его не беспокоили (его жена, конечно, была), но при ходьбе чувствовал себя неуравновешенным. Стало очевидно, что он фактически терял равновесие и память в течение примерно 12 месяцев, но за последние 2 месяца это прогрессировало быстрее. У него не было ни головных болей, ни сенсорных симптомов, ни симптомов мочеиспускания. Он хорошо спал и ел, не похудел.Он был опытным пианистом, и его музыкальные способности не ухудшились. Он никогда не курил и мало пил.
Экзамен
Его жена сказала, что он был его обычным шутником — возможно, даже глупым. Общий осмотр, включая сердце, грудную клетку и живот, был нормальным. Не было аномалий черепных нервов, отека зрительного нерва, мышечной атрофии, нормального мышечного тонуса и силы во всем; в верхних конечностях рефлексы в норме, коленные и голеностопные рефлексы отсутствуют.Его правая подошва была разгибательной. Ощущение не пострадало от булавочного укола; было небольшое нарушение вибрации и ощущения положения пальцев ног. Он ходил широкой неустойчивой походкой и не мог ходить тандемом; Проба Ромберга была положительной. Он набрал 22/30 на кратком экзамене по психическому состоянию, потеряв баллы за ориентацию, память (вспоминание) и повторение. На когнитивном экзамене Адденбрука он набрал 62/100, потеряв баллы по памяти (отзыв, антероградный и ретроградный). Снижена беглость речи.Язык, наименование и понимание остались нетронутыми. Его оценки были следующими: внимание и ориентация 14/18, память 10/26, беглость 0/14, язык 24/26, зрительно-пространственное восприятие 14/16.
МРТ головного мозга (рис. 1), который он принес с собой, показал следующее: «Имеется умеренная церебральная атрофия с выступом кортикальных борозд и желудочковой системы с расширением тел и височных рогов боковых желудочков. третьего желудочка. Четвертый желудочек виден.Повышенная интенсивность сигнала в перивентрикулярном белом веществе указывает на резорбцию перивентрикулярной спинномозговой жидкости (ЦСЖ). В большой цистерне имеется скопление спинномозговой жидкости, деформирующее нижние части полушарий мозжечка, что указывает на арахноидальную кисту. На изображениях нет ишемии мелких сосудов. Дистальная левая позвоночная артерия окклюзирована. Комментарий: несмотря на церебральную атрофию, по внешнему виду можно предположить гидроцефалию с нормальным давлением ».
МРТ головного мозга при предъявлении.Последовательности восстановления инверсии осевого T2 (A, B) и корональной жидкости показывают атрофию коры и увеличение желудочков с возможной экссудацией перивентрикулярной спинномозговой жидкости.
В отчете МРТ брюшной полости описаны «четыре кисты в поджелудочной железе без увеличения, масс-эффекта, окружающей инфильтрации или расширения протока».
Обсуждение, часть A (д-р GM Halmagyi)
У этого пациента три различных клинических синдрома: деменция (нарушение памяти и беглости речи), атаксия (широкая походка и положительный результат теста Ромберга) и сенсорная нейропатия (отсутствие рефлексов коленного и голеностопного суставов с нарушением чувствительности в дистальных отделах).Атаксия может быть вызвана сенсорными нарушениями, корковыми нарушениями или не связанной причиной, например, мозжечковыми или вестибулярными нарушениями. Темп деменции и атаксии подострый; темп невропатии неизвестен. Во вставке 1 подробно описан возможный диагноз, который следует учитывать. Какое заболевание могло вызвать все три этих синдрома? Возможные состояния включают дефицит витамина B1 (Вернике – Корсакова), дефицит витамина B12 (подострая комбинированная дегенерация спинного мозга) и паранеопластические заболевания.
Дефицит витамина B12 объяснил бы все неврологические проблемы1, 2, а низкий уровень витамина B12 в сыворотке подтвердил бы диагноз, даже без гематологических изменений. Неврологический дефицит B12 может возникать даже при нормальном уровне B12 в сыворотке3, особенно у пациентов, подвергшихся воздействию метформина4 или закиси азота (в медицинских или рекреационных целях) .5 В таких случаях высокие уровни гомоцистеина или метилмалоновой кислоты в сыворотке крови — промежуточных метаболитов B12 — указывают на функциональный дефицит B12. .6 Изменения МРТ головного мозга могут произойти при дефиците B123, хотя изменения МРТ позвоночника более обычны.
Синдром Вернике – Корсакова в контексте хронического злоупотребления алкоголем (для объяснения периферической невропатии) 7 также объясняет все неврологические нарушения, даже без офтальмоплегии. Однако пациентка хорошо питалась и употребляла мало алкоголя, что делает этот диагноз маловероятным. Энцефалопатия Вернике имеет характерные аномалии МРТ в стволе мозга и базальных ганглиях, включая гиперинтенсивность Т2 вокруг третьего желудочка, в дорсомедиальных областях таламуса, периакведуктальных областях среднего мозга и мамиллярных телах.8
Паранеопластические расстройства . Паранеопластический лимбический энцефалит обычно проявляется когнитивными изменениями, включая ухудшение кратковременной памяти, судороги, поведенческие и личностные изменения; 9 могут быть антитела к Hu, CV2, Ma2 или потенциалзависимым калиевым каналам (VGKC) 9. связаны с паранеопластической дегенерацией мозжечка и сенсорной нейропатией, 10 что может объяснить нарушение походки у этого пациента. На МРТ мезиальные височные структуры могут быть опухшими и гиперинтенсивными на Т2-взвешенных последовательностях.11 У этого пациента не было изменений поведения и личности, психических симптомов или судорог, а отсутствие типичных аномалий МРТ снижает вероятность лимбического энцефалита.
Для дальнейшего исследования возможности лимбического энцефалита нам понадобится спинномозговая жидкость, ищущая легкий лейкоцитоз и повышенный уровень белка.9 ЭЭГ может показывать очаговое или генерализованное замедление или даже максимальную эпилептиформную активность в височных областях.12 Нам также необходимо провести тест на антинейрональные антитела. и антитела к VGKC.Затем ему требуется визуализация для выявления подлежащей опухоли, включая рентген грудной клетки, компьютерную томографию / позитронно-эмиссионную томографию (ПЭТ) всего тела, а также маркеры опухолей сыворотки (карциноэмбриональный антиген (CEA), Ca19.9, α-фетопротеин).
Коробка 1Дифференциальный диагноз для этого пациента включает
Гидроцефалия : сообщающийся (включая гидроцефалию нормального давления или инфильтрацию мозговых оболочек в результате инфекции), не сообщающийся.
Новообразование : интрапаренхиматозное и / или менингитное.
Паранеопластическое состояние : например, новообразование, продуцирующее антитела против Hu, вызывающее когнитивный дефицит и атаксию, или потенциал-зависимые калиевые каналы и энцефалит против рецепторов NMDA.
Инфекционный энцефалит : например, вирусный энцефалит, инфекционный менингит (туберкулезный, криптококковый), ВИЧ-энцефалит, болезнь Уиппла, сифилис.
Аутоиммунный : например, саркоидоз.
Спорадическая болезнь Крейтцфельдта – Якоба
Токсический метаболизм : например, энцефит Хашимото, дефицит витамина B1, дефицит витамина B12, гипонатриемия и синдром несоответствующей секреции антидиуретического гормона.
Сосудистые причины : церебральный васкулит, тромбоз вен головного мозга, ишемия мелких сосудов
Заболевания белого вещества с поздним началом : например, наследственная диффузная лейкоэнцефалопатия с нейроаксональными сфероидами.
Лекарственные средства : нейролептики, бензодиазепины, рекреационные наркотики.
Быстрое прогрессирование хронического нейродегенеративного расстройства: Болезнь Альцгеймера, лобно-височная деменция.
Среди паранеопластических причин следует рассмотреть энцефалит против рецепторов N-метил-D-аспартата (NMDAR). Этот характерный синдром развивается в три этапа. Первый — это продромальное заболевание с лихорадкой, головной болью, тошнотой, желудочно-кишечными или респираторными симптомами. Далее следуют психиатрические симптомы, проблемы с кратковременной памятью и речью. На третьей фазе у пациентов развиваются снижение отзывчивости, аномальные движения (обычно оролингвальные дискинезии лица) и вегетативные симптомы.В половине случаев результаты МРТ являются аномальными, с повышенной интенсивностью сигналов T2 или FLAIR в гиппокампе, мозжечке или коре головного мозга, лобно-базальных и островковых областях, базальных ганглиях, стволе головного мозга и, реже, в спинном мозге. СМЖ отклоняется от нормы в 80% случаев; включая лимфоцитарный плеоцитоз и нормальный или с умеренно повышенным содержанием белка. В 60% случаев присутствуют олигоклональные полосы, специфичные для спинномозговой жидкости. 13 Возраст этого пациента не соответствует анти-NMDAR-энцефалиту, его средний возраст составляет 19 лет. Однако в некоторых случаях возраст пациентов превышает 70 лет.13 Тестирование на антитела к рецептору NMDA (N-метил-D-аспартат) помогло бы исключить этот диагноз.
При возможном паранеопластическом заболевании следует учитывать другие возможные причины путаницы, такие как гипонатриемия (из-за синдрома несоответствующей секреции антидиуретического гормона) и гиперкальциемия. Следовательно, нам нужно будет увидеть его сывороточные электролиты, осмоляльность и сывороточный кальций с поправкой на уровни сывороточного альбумина.
Если мы предположим, что периферическая нейропатия является хронической и диабетической и не связана с его проявлением, что тогда может вызвать только подострую деменцию и атаксию? Дифференциальный диагноз становится намного шире.Однако сначала мы должны рассмотреть возможность того, что деменция и атаксия на самом деле вызваны гидроцефалией, как это показано на МРТ.
Гидроцефалия
Увеличение желудочков возникает либо из-за проблемы с абсорбцией спинномозговой жидкости (гидроцефалия), либо из-за сокращения ткани головного мозга (церебральная атрофия) при нейродегенеративных заболеваниях и нормальном старении. На практике иногда бывает сложно определить причину увеличения желудочков. Гидроцефалия либо коммуникативная, либо не коммуникативная (т. Е. Обструктивная).
Обструктивная гидроцефалия означает блокировку оттока спинномозговой жидкости в желудочковой системе. Без закупоривающей массы это часто вызвано стенозом водопровода, соединяющего 3-й и 4-й желудочки: следовательно, 4-й желудочек не увеличен.
Сообщающаяся гидроцефалия означает, что CSF свободно проходит через желудочковую систему и выходит из четвертого желудочка через отверстия Люшки и Мажанди.Проблема обычно заключается в нарушении циркуляции или абсорбции спинномозговой жидкости в субарахноидальном пространстве. При сообщающейся гидроцефалии увеличивается вся желудочковая система, включая 4-й желудочек. Сообщающаяся гидроцефалия может возникать остро или хронически у пациентов с менингитом или субарахноидальным кровоизлиянием.
Особую разновидность хронической коммуникативной гидроцефалии у пожилых людей называют «нормальным давлением», потому что давление спинномозговой жидкости при случайной одиночной люмбальной пункции является нормальным.Это вызывает серьезные диагностические трудности у пожилых пациентов с деменцией, которые также не имеют равновесия и страдают недержанием, поскольку эта конкретная триада неврологических проблем вместе с увеличенной желудочковой системой может быть вызвана нейродегенеративным заболеванием с церебральной атрофией, а не гидроцефалией при нормальном давлении НПХ) .14
Могут ли проблемы этого пациента быть следствием (коммуникативной) гидроцефалии, как предполагает отчет МРТ головного мозга, и может ли это быть нормальное давление? Хотя его клинический синдром (деменция + атаксия) согласуется с НПХ, у большинства пациентов с деменцией и атаксией НПХ нет, даже если у них есть третий компонент триады НПХ (недержание).14 Исследования, которые просто документируют нарушение походки, деменцию, недержание мочи и вентрикуломегалию, вероятно, переоценивают распространенность НПХ, которая, вероятно, равна распространенности прогрессирующего надъядерного паралича. , внимание и скорость психомоторности.15 Нейропсихологический профиль может улучшиться после установки шунта.16 Однако другие исследования показали, что, хотя походка может улучшаться на начальном этапе, недержание мочи и когнитивные функции не улучшаются после установки шунта, что свидетельствует о высокой степени гипердиагностики.14 Не существует окончательного теста на НПХ. Существует несколько дополнительных тестов для помощи в диагностике, в том числе «спин-тест спинномозговой жидкости» (люмбальная пункция большого объема), наружный дренаж спинномозговой жидкости с помощью спинномозгового дренажа и определение оттока спинномозговой жидкости с помощью исследований соответствия инфузии спинномозговой жидкости.17 Однако эти тесты имеют разную чувствительность и специфичность. . Тест спинного мозга имеет низкую чувствительность 26–61%. Клинический ответ на удаление спинномозгового катетера имеет более высокую чувствительность 50–100%, специфичность 60–100% и прогностическую ценность положительного результата 80–100%.18 Однако это инвазивная процедура, требующая квалифицированного управления и высокая частота осложнений.
Скорость прогрессирования у этого пациента в течение нескольких недель кажется слишком высокой для НПХ, который обычно развивается в течение месяцев или лет. Итак, если действительно имеется сообщающаяся гидроцефалия, но не при нормальном давлении, что еще может быть ее причиной? Придется подумать об инфекционных или неопластических причинах.
Инфекционные и неопластические причины
Многие инфекции ЦНС могут вызывать снижение когнитивных функций.Результаты CSF помогают дифференцировать их, включая количество белка и лейкоцитов в CSF и вирусную ПЦР (вирус простого герпеса (HSV), цитомегаловирус (CMV), вирус Эпштейна-Барра (EBV) и энтеровирус). Однако у нашего пациента этому противостоит относительно подострое начало и отсутствие системных признаков, включая лихорадку. Однако нейротропные вирусы, такие как энтеровирус 71, могут проявляться подострым снижением когнитивных функций, которое может быть фатальным.19 Кроме того, корь может вызывать снижение когнитивных функций у пожилых пациентов20, а микоплазма может вызывать энцефалит.21 У пациентов с ослабленным иммунитетом инфекции ЦМВ и ВЭБ могут вызывать энцефалит22, поэтому важно знать ВИЧ-статус пациента. Болезнь Лайма также может иметь слабоумие.
Хронический менингит, вызванный криптококковой или туберкулезной инфекцией или лептоменингеальными метастазами, может проявляться деменцией и атаксией и вызывать внутричерепную гипертензию с (например, гидроцефалией) 24, 25 или без увеличения желудочков (pseudotumour cerebri) 26.
Примерно в половине случаев злокачественный менингит проявляется головной болью, слабоумием и атаксией.27 Менингеальная лимфома может вызвать повышение внутричерепного давления с отеком папилломы и / или гидроцефалией. 28 Рак груди (3%), мелкоклеточный рак легкого (6%), немелкоклеточный рак легкого (1%), опухоли желудочно-кишечного тракта (0,015– 0,25%), первичная карцинома неизвестного происхождения (3%) и меланома (1,5%) могут вызывать неопластический менингит, 27 и могут вызывать повышение внутричерепного давления и гидроцефалию из-за снижения абсорбции спинномозговой жидкости паутинными ворсинками. У этого пациента «кисты поджелудочной железы», выявленные на МРТ, могут представлять собой возможное неопластическое поражение.Быстрый клинический спад также согласуется с этим диагнозом.
На МРТ можно было бы искать лептоменингеальное усиление. Было бы важно знать давление спинномозговой жидкости в поясничном отделе, белок, глюкозу и дифференциальное количество лейкоцитов, а также данные цитологии и проточной цитометрии для обнаружения любых злокачественных клеток. Содержание белка в спинномозговой жидкости и количество лейкоцитов могут быть наиболее полезными тестами для дифференциации вирусного, бактериального и криптококкового менингита, в то время как концентрация глюкозы в спинномозговой жидкости менее полезна.29 Низкий уровень глюкозы указывает на метаболически активный процесс, но уровень глюкозы может быть низким как при инфекционных, так и опухолевых заболеваниях. менингит.Также было бы важно знать результаты ПЦР на криптококковый антиген и туберкулез (ТБ) на спинномозговую жидкость и серологические анализы на криптококки в крови. Для диагностики злокачественного менингита может потребоваться несколько исследований спинномозговой жидкости.
Может ли это быть нейросифилис в одной из его многогранных форм? Диагноз зависит от результатов серологических тестов крови и спинномозговой жидкости; интерпретация этих результатов часто затруднена — положительный результат может не означать, что причиной является сифилис. В первую очередь следует проверить серологию крови на сифилис и, в случае положительного результата, исследовать СМЖ.Диагноз следует рассматривать, если трепонемный тест дает положительный результат в спинномозговой жидкости, имеется повышенное количество мононуклеарных клеток или положительный результат референс-лаборатории венерических заболеваний / экспресс-реактива плазмы (VDRL / RPR) на спинномозговой жидкости. В то время как положительный трепонемный тест на спинномозговую жидкость не подтверждает активный нейросифилис, отрицательный тест исключает его.
Возможна также неврологическая болезнь Уиппла. У пациентов наблюдается снижение когнитивных функций, психические симптомы, судороги и окулопалатальный миоклонус.МРТ должна показать одно или несколько многоузловых очагов поражения. Серологический анализ спинномозговой жидкости на Tropheryma whipplei не всегда положительный, и для постановки диагноза может потребоваться биопсия головного мозга.31
Следует также учитывать прогрессирующую мультифокальную лейкоэнцефалопатию. Обычно у пациентов наблюдаются слабость, сенсорные изменения, гемианопсия, снижение когнитивных функций и дисбаланс, а также трудности с походкой. МРТ имеет решающее значение для диагностики, обычно выявляя области гиперинтенсивности на изображениях T2 и FLAIR и гипоинтенсивность на T1.Обычно вовлекаются подкорковое белое вещество, ножки мозжечка, базальные ганглии и таламус.32 ЦСЖ следует проверять на ДНК вируса JC.
Болезнь Крейтцфельдта-Якоба
Учитывая скорость снижения, когнитивных нарушений и нарушений походки, важно учитывать спорадическую болезнь Крейтцфельдта – Якоба (sCJD). Это может привести к когнитивным (39%), мозжечковым (29%), поведенческим (20%), конституциональным (20%), сенсорным (11%), моторным (9%) и зрительным (7%) симптомам.33, 34 Чтобы соответствовать диагностическим критериям ВОЗ для CJD, пациентам необходимы два из следующего: миоклонус, пирамидные / экстрапирамидные признаки, зрительный или мозжечковый дефицит и акинетический мутизм.У этого пациента был подошвенный разгибатель справа, что соответствовало пирамидальной находке, и проблема походки могла представлять собой дефицит мозжечка. Результат ЭЭГ был бы важен, особенно при поиске периодических эпилептиформных разрядов. Также CSF ищет белок 14-3-3 и нейрон-специфичную енолазу. На спинномозговой жидкости можно было бы также ожидать умеренно повышенного уровня белка, нормального уровня глюкозы и отсутствия лейкоцитоза.35 При sCJD диффузно-взвешенная (DWI) гиперинтенсивность в корковом и подкорковом сером веществе высокочувствительна и специфична и превосходит либо ЭЭГ, либо нейроны спинномозговой жидкости. специфическая енолаза или белок 14-3-3.36
Токсические / метаболические причины
Мы также должны учитывать токсическую / метаболическую причину. Энцефалопатия Хашимото может проявляться снижением когнитивных функций и атаксией, хотя, как правило, рецидивирующе-ремиттирующим течением и приступами, подобными инсульту, или судорогами.37 Было бы важно знать результаты функции щитовидной железы и результаты антител к щитовидной железе. Снижение когнитивных функций из-за периодического воздействия окиси углерода38 может проявляться замедлением и ухудшением памяти, наряду с аномалиями походки39 и (в отличие от этого) с аномалиями бледного шара на МРТ.40
Церебральный васкулит
Изолированный церебральный васкулит может проявляться подострым снижением когнитивных функций и неспецифическими симптомами. Диагноз сложно поставить и полагается на исследование спинномозговой жидкости, МРТ / магнитно-резонансную ангиографию и цифровую вычитающую ангиографию. И МРТ, и ангиография могут казаться нормальными и иметь низкую диагностическую специфичность. Для постановки диагноза часто требуется биопсия головного мозга41.
Церебральный венозный тромбоз
Церебральный венозный тромбоз как глубоких, так и поверхностных пазух может проявляться подострой с неспецифическими признаками и изменением мышления и познания.Снижение когнитивных функций может быть следствием как изменений белого вещества, так и венозной гипертензии, что приводит к повышению внутричерепного давления. 42 Было бы важно просмотреть МРТ и МР-венограмму пациента в поисках тромба и измерить давление открытия спинномозговой жидкости.
Заболевания белого вещества с поздним началом
Лейкодистрофии у взрослых могут проявляться подострым снижением когнитивных функций, проблемами походки и атаксией. В частности, метахроматическая лейкодистрофия, адренолейкодистрофия, болезнь исчезающего белого вещества и наследственная диффузная лейкоэнцефалопатия с аксональными сфероидами (ЛПВП) часто сопровождаются ухудшением психического состояния и когнитивными изменениями.43, 44 Однако отсутствие изменений белого вещества на МРТ, спастичность или другие неврологические признаки делают лейкодистрофию маловероятной, хотя когнитивное снижение может предшествовать изменениям МРТ в HDLS.
Аутоиммунное заболевание
Мы должны учитывать аутоиммунные причины, такие как саркоидоз или синдром Шегрена. Могут помочь анализы крови, включая аутоиммунный и васкулитный скрининг, а также рентген грудной клетки при внутригрудной лимфаденопатии. МРТ может показать изменения белого вещества и вовлечение менингеальных сосудов.
Наркотики
Наркотики могут вызывать снижение когнитивных функций.В частности, нейролептики могут вызывать быстрое снижение когнитивных функций у пациентов с деменцией с тельцами Леви. Однако у этого пациента не было галлюцинаций, нарушений поведения во сне в фазе быстрого сна или экстрапирамидных особенностей. Несмотря на это, было бы важно дважды проверить историю приема лекарств, чтобы убедиться, что он не получал нейролептики.
Хронические нейродегенеративные болезни
Хронические нейродегенеративные заболевания могут проявляться подостро и быть ошибочно приняты за быстро прогрессирующую деменцию.В ретроспективном исследовании у 23% пациентов с диагнозом быстро прогрессирующая деменция была лобно-височная деменция — заболевание двигательных нейронов, а у 9% — болезнь Альцгеймера (45). У этого пациента в течение 12 месяцев наблюдалось снижение, за которым следовали еще 2 месяца. быстрое ухудшение состояния; возможно, у него была ранняя болезнь Альцгеймера. В этой ситуации было бы важно выполнить полный септический скрининг (кровь, рентген грудной клетки и моча), чтобы убедиться, что не пропущена инфекция, усугубляющая хроническое заболевание.Тщательное исследование МРТ и объемного МРТ может быть полезным при поиске очаговой атрофии, которая может указывать на болезнь Альцгеймера или лобно-височную деменцию.
Требуются анализы
Чтобы обследовать этого пациента на основе списка дифференциальных диагнозов, мне нужны результаты следующих тестов.
Кровь: общий анализ крови, электролиты, тесты функции печени, тесты функции щитовидной железы, антитела к щитовидной железе, аутоиммунный и васкулитный скрининг, витамин B12 и фолат, гомоцистеин, метилмалоновая кислота, антинейрональные антитела, антитела к VGKC, антитела к рецепторам NMDA, серология сифилиса и онкомаркеры (CEA, α-фетопротеин, Ca19.9)
Визуализация: повторная МРТ головного мозга с контрастированием, рентген грудной клетки, ПЭТ / КТ-сканирование всего тела.
CSF: давление, количество лейкоцитов, белок микроскопии, глюкоза, олигоклональные полосы, TB, Whipple, HSV, CMV, EBV и энтеровирусная ПЦР, криптококковый антиген, цитология, проточная цитометрия и VDRL.
ЭЭГ.
Патология (доктор М. Родригес)
Произведено полное вскрытие. На верхушке нижней доли правого легкого обнаружена умеренно высокодифференцированная аденокарцинома диаметром 4 см.Правые прикорневые лимфатические узлы были увеличены, слиплись и содержали метастатическую карциному. Образований надпочечников не было.
Мозг весил 1190 г. Стеноз дистального отдела позвоночной артерии атеромой и ретроцеребеллярной арахноидальной кистой составил> 90%. Отверстия Magendie и Luschka были проходимыми. Наблюдались утолщение и изменение цвета базальных лептоменингов на желтый / оранжевый. Была гидроцефалия с симметричным расширением бокового и третьего желудочков (рис. 4), а также увеличением водопровода и четвертого желудочка.Пеллюцидная перегородка не была ни фенестрирована, ни истончена. Около желудочков белое вещество было бледным и мягким.
Рисунок 4Коронарный разрез головного мозга на уровне ядер латеральных коленчатых стволов, демонстрирующий заметное симметричное увеличение латерального и третьего желудочков. Бледность белого вещества, прилегающего к желудочкам, возникает из-за перивентрикулярного отека, вторичного по отношению к трансепендимному распространению спинномозговой жидкости.
Микроскопически, лептоменингеальный карциноматоз широко распространен почти в каждом исследованном отделе с распространением в пространства Вирхова – Робина, и было несколько микроскопических очагов поверхностной паренхиматозной инвазии.Опухоль напоминала аденокарциному легких (рис. 5А, В). Опухоль также инфильтрировала экстрааксиальные сегменты нескольких черепных нервов и дорсальные и вентральные корешки спинного мозга (рис. 5C, D), наиболее заметные в поясничной области, где несколько нейронов в вентральных рогах демонстрируют центральный хроматолиз. Также было несколько областей недавнего инфаркта, преимущественно в коре головного мозга, часто связанных с инфильтрацией опухоли в пространства Вирхова-Робина.
Рисунок 5.(A) Умеренно хорошо дифференцированная аденокарцинома в субарахноидальном пространстве (H&E).Исходное увеличение × 100 (B) Аденокарцинома в субарахноидальном пространстве, расширяющаяся в пространство Вирхова-Робинса (H&E). Исходное увеличение × 400 (C) Поясничный отдел спинного мозга с лептоменингеальным карциноматозом с инфильтрацией дорсального и вентрального корешков. × 12,5 (иммунопероксидаза цитокератин 8). Оригинальное увеличение × 12,5. (D) Поясничные вентральные корешки, показывающие инфильтрацию опухоли (цитокератин 8 иммунопероксидаза). Оригинальное увеличение × 400.
Легкое заболевание мелких сосудов белого вещества головного мозга не было достаточно серьезным, чтобы объяснить МРТ или клинические особенности.Была широко распространена активация микроглии, но не было воспаления, подтверждающего диагноз васкулита, паранеопластического или аутоиммунного энцефалита. Также отсутствовали признаки инфекции, прионной болезни, острой или хронической энцефалопатии Вернике, подострой комбинированной дегенерации или хронического нейродегенеративного заболевания, включая лобно-височную долевую дегенерацию, болезнь Альцгеймера или слабоумие с болезнью тельцов Леви. Гидроцефалия, скорее всего, возникла из-за нарушения оттока спинномозговой жидкости в субарахноидальном пространстве, вторичного по отношению к лептоменигальному карциноматозу, вызванному аденокарциномой правого легкого.
Заключительные замечания
У этого пациента были деменция, атаксия и сообщающаяся гидроцефалия из-за злокачественного менингита, вызванного первичной аденокарциномой легкого. Неопластический менингит лишь в редких случаях возникает в результате немелкоклеточного рака легкого и встречается только у 1% пациентов27. В серии из 640 пациентов со злокачественным менингитом только 37 имели гидроцефалию47.
Ключом к диагностике неопластического менингита является МРТ головного и спинного мозга с контрастным усилением и цитология спинномозговой жидкости.В исследовании 137 пациентов с клиническими симптомами неопластического менингита визуализация была аномальной у 55%, включая усиление лептоменингеального, субэпендимального, твердого мозга или черепно-мозговых нервов; поверхностные церебральные поражения или сообщающаяся гидроцефалия.48 Однако МРТ головного мозга с контрастным усилением может не дать положительного результата. Недавнее исследование 44 пациентов с неопластическим менингитом обнаружило чувствительность 45% для выявления менингеального поражения: чувствительность была выше для солидных опухолей (84,6%) и ниже для лейкемии (20%) и лимфомы (37.5%) .49 В этом случае ни МРТ позвоночника, ни МРТ головного мозга с контрастным усилением не выполнялись, но тот факт, что мозг и позвоночник были макроскопически нормальными при вскрытии, предполагает, что однозначного увеличения контрастности не было. Возможно, что у пациента действительно появились очаговые признаки, указывающие на вовлечение позвоночника в конце его болезни, но к тому времени он был слишком ослаблен, чтобы обследоваться клинически. Однако есть сообщения о случаях злокачественных новообразований, особенно лимфомы, с лептоменингеальным поражением корешков спинномозговых нервов, которые не имеют локализующих признаков 28, а в некоторых случаях — с нормальной визуализацией позвоночника.50
Чувствительность цитологии спинномозговой жидкости при злокачественном менингите зависит от объема образца и времени до фиксации. 46 У этого пациента признаками менингеального процесса были высокое содержание белка и низкое содержание глюкозы, но цитология не показала злокачественных клеток. Оптимальный метод цитологического исследования спинномозговой жидкости — это взять не менее 10 мл спинномозговой жидкости и собрать образец в пробирку с фиксатором формалина перед отправкой в лабораторию.51 Цитология имеет чувствительность 93% для выявления неопластического менингита.