Деменция пресенильная: ФГБНУ НЦПЗ. ‹‹Клиника приобретенного слабоумия››
ФГБНУ НЦПЗ. ‹‹Эндогенно-органические психические заболевания››
Форма обратной связи
Вопрос по работе сайтаВопрос специалистуВопрос в администацию клиники
Адрес email
Имя
Текст сообщения
Современная международная классификация деменций альцгеймеровского типа предельно проста и основана на возрастном принципе.
В соответствии с МКБ-10 (1994) выделяются две формы: 1) болезнь Альцгеймера с ранним началом, т. е. до 65 лет (син.: тип 2 болезни Альцгеймера, пресенильная деменция альцгеймеровского типа). Эта форма соответствует классической болезни Альцгеймера и в литературе иногда обозначается как «чистая» (pure) болезнь Альцгеймера; 2) болезнь Альцгеймера с поздним, т. е. после 65 лет началом (син.: тип 1 болезни Альцгеймера, сенильная деменция альцгеймеровского типа). Предусмотрено также выделение атипичной болезни Альцгеймера или деменций смешанного типа, т.
По существу тот же хронологический принцип заложен и в DSM-IV (1994), где в рамках каждой из двух основных форм деменций альцгеймеровского типа — с ранним (до 65 лет) и поздним (после 65 лет) началом предусмотрено выделение вариантов, характеризующихся наличием в клинической картине (помимо синдрома деменций) различных продуктивных психопатологических расстройств. В соответствии с этим выделены варианты ранней и поздней деменций альцгеймеровского типа с делирием, с бредом, с депрессивными расстройствами и без таковых — неосложненные.
Приведенные классификационные схемы, основанные на формально возрастном и отчасти феноменологических критериях, несомненно, отличаются простотой и удобством использования в практической медицине. Однако они вряд ли могут считаться удовлетворительными с точки зрения этиологически или патогенетически ориентированного подхода. В этом отношении более адекватной представляется классификация шведских исследователей [Wallin A.
Описание указанными авторами клинических различий болезни Альцгеймера и сенильной деменций альцгеймеровского типа совпадает с основными дифференцирующими признаками для разграничения этих форм деменций, разработанными в отечественной геронтопсихиатрии [Гаврилова С. И. и др., 1992]. Они приводятся в табл.
Классификация, основанная на своеобразии клинических проявлений и течения, характерных для различных форм деменций альцгеймеровского типа, а не только на различиях в возрасте начала болезни, представляется более адекватной еще и потому, что из-за медленного малозаметного прогрессирования заболевания на начальном этапе его развития истинный возраст больного к его началу бывает определить трудно, а порой даже невозможно. Кроме того, небольшая часть случаев «классической» болезни Альцгеймера может начинаться в возрасте, превышающем 65 лет, а инициальные симптомы сенильной деменций, напротив, в отдельных случаях становятся очевидными до 65 лет.
Таблица 1. Параметры, применяемые в дифференциации основных клинических форм деменций альцгеймеровского типа
Болезнь Альцгеймера (пресенильная деменция альцгеймеровского типа) | Сенильная деменция альцгеймеровского типа |
Начало преимущественно в пресенильном возрасте | Начало преимущественно в старческом возрасте |
Медленное развитие болезни на инициальных этапах и бурное прогрессирование на этапе клинически выраженной деменций | Менее прогредиентное развитие болезни на всех этапах (за исключением конечного) |
Появление корковых очаговых расстройств уже на ранних этапах болезни | Нарушение высших корковых функций на фоне далеко зашедшей деменций |
Множественное тяжелое поражение высших корковых функций на этапе продвинутой деменций | Общее ухудшение высших корковых функций, которое редко достигает степени явных очаговых расстройств |
Длительная сохранность реакции пациента на болезнь и основных его личностных особенностей | Выраженные изменения личности и утрата критики к болезни уже на ранних ее этапах |
| Относительно гомогенная клиническая картина на развернутом этапе деменций (афато-апракто-агностическая деменция) | Гетерогенная клиническая картина (различные клинические формы) деменций |
Чрезвычайно важно как в клинической практике, так и для исследовательских целей адекватно и единообразно оценивать заболевание в зависимости от стадии его развития, что в случае деменций альцгеймеровского типа равноценно по существу тяжести деменций. Наиболее адекватной в этом отношении нам представляется шкала, разработанная американскими исследователями [Hughes С. P. et al., 1982] — Clinical Dementia Rating (CDR), позднее усовершенствованная L. Berg (1984, 1988). В этой шкале предусматривается выделение четырех последовательных стадий развития болезни Альцгеймера — от стадии сомнительной деменций (CDR-0,5) через стадию мягкой (CDR-1) и умеренной (CDR-2) до тяжелой (CDR-3) деменций, при этом нулевая оценка соответствует отсутствию когнитивных нарушений и изменений в уровне социальной и профессиональной деятельности. Описываемая шкала основана на оценке функциональных возможностей пациентов на каждом из последовательных этапов деменций и в целом соответствует принятому в отечественной геронтопсихиатрии выделению этапов развития деменций альцгеймеровского типа [Калын Я. Б., 1990; Селезнева Н. Д., 1990].
Наиболее адекватной в этом отношении нам представляется шкала, разработанная американскими исследователями [Hughes С. P. et al., 1982] — Clinical Dementia Rating (CDR), позднее усовершенствованная L. Berg (1984, 1988). В этой шкале предусматривается выделение четырех последовательных стадий развития болезни Альцгеймера — от стадии сомнительной деменций (CDR-0,5) через стадию мягкой (CDR-1) и умеренной (CDR-2) до тяжелой (CDR-3) деменций, при этом нулевая оценка соответствует отсутствию когнитивных нарушений и изменений в уровне социальной и профессиональной деятельности. Описываемая шкала основана на оценке функциональных возможностей пациентов на каждом из последовательных этапов деменций и в целом соответствует принятому в отечественной геронтопсихиатрии выделению этапов развития деменций альцгеймеровского типа [Калын Я. Б., 1990; Селезнева Н. Д., 1990].
Пресенильные деменции: болезнь Альцгеймера и Пика
Пресенильные деменции — сборная группа развивающихся в предстарческом возрасте состояний слабоумия, обусловленных атрофией мозга. В рамках пресенильных деменции выделяют ряд самостоятельных нозологических форм, среди которых ведущее место с учетом их большей встречаемости принадлежит болезням Альцгеймера и Пика. Другие заболевания, относящиеся к пресенильным деменциям (хорея Гентингтона, болезнь Паркинсона), диагностируются очень редко.
В рамках пресенильных деменции выделяют ряд самостоятельных нозологических форм, среди которых ведущее место с учетом их большей встречаемости принадлежит болезням Альцгеймера и Пика. Другие заболевания, относящиеся к пресенильным деменциям (хорея Гентингтона, болезнь Паркинсона), диагностируются очень редко.
Болезнь Альцгеймера
Заболевание описано А. Альцгеймером в 1906 г. Оно развивается в возрасте 45—65 лет, чаще всего в 55—60 лет.
Дебют болезни медленный, течение — прогредиентное. Ослабоумливающий процесс начинается с мнестических нарушений в виде ослабления памяти на текущие события, которое постепенно переходит в фиксационную амнезию, а затем в прогрессирующую амнезию. Возникает глубокая амнестическая дезориентировка. В течение нескольких лет почти полностью опустошаются запасы знаний и умений. В отличие от старческой деменции для болезни Альцгеймера не характерны конфабуляции, сдвиг ситуации в прошлое.
Параллельно с расстройствами памяти нарастают нарушения мышления.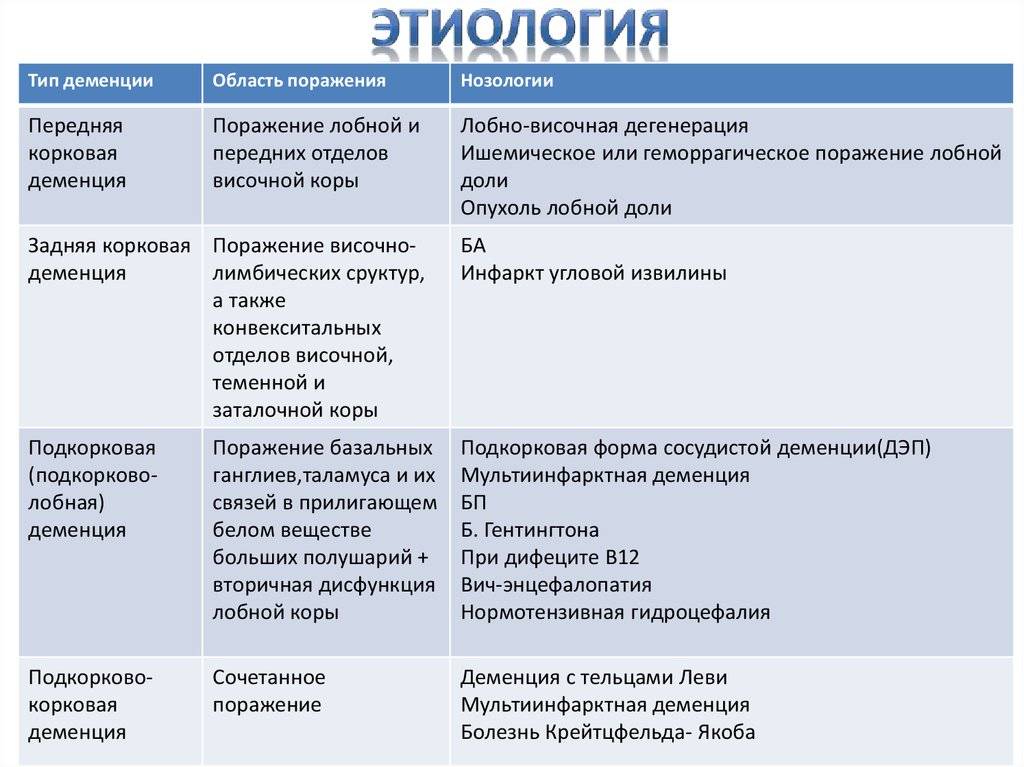 Они начинаются с затруднений в более сложной аналитико-синтетической деятельности, а заканчиваются полной интеллектуальной беспомощностью. В итоге выявляется глубокое слабоумие.
Они начинаются с затруднений в более сложной аналитико-синтетической деятельности, а заканчиваются полной интеллектуальной беспомощностью. В итоге выявляется глубокое слабоумие.
На ранних этапах болезни нередко возникают продуктивные психотические расстройства в виде мелкомасштабного бреда ущерба, отравления, ревности, реже — в форме галлюциноза. У многих больных отмечаются эпилептиформные припадки.
Одна из особенностей болезни Альцгеймера по сравнению с сенильной деменцией состоит в том, что на ее начальной стадии сохраняется сознание собственной несостоятельноста, болезненной измененности, которому сопутствуют тревога и растерянность, позднее сменяющиеся безучастностью, полным равнодушием.
Самое существенное клинические своеобразие болезни Альцгеймера заключается в сочетании усиливающейся интеллектуально-мнестической недостаточности с нарастающими расстройствами высших корковых функций — речи, чтения, письма, счета, гнозиса и праксиса.
Первые признаки неврологических нарушений корковых функций часто удается обнаружить уже на ранних этапах заболевания (С. И. Гаврилова). Они выражаются в затрудненном осмыслении чужой речи, в нечеткости произношения, затруднениях и ошибках при письме, чтении, счете, забывании названий отдельных предметов. Указанные нарушения интенсивно прогрессируют и сменяются сенсорной, амнестической и агностической афазией. Речь становится все более дизартричной, все большее место в ней занимают стереотипные речевые обороты, междометия, вводные слова. Происходят задержки при произнесении начальных букв и слогов, многократное повторение их (логоклоническое заикание). В дальнейшем активная речь ограничивается бессмысленным повторением обрывков слов или отдельных звуков. Почти полностью утрачиваются способности чтения, письма, счета.
И. Гаврилова). Они выражаются в затрудненном осмыслении чужой речи, в нечеткости произношения, затруднениях и ошибках при письме, чтении, счете, забывании названий отдельных предметов. Указанные нарушения интенсивно прогрессируют и сменяются сенсорной, амнестической и агностической афазией. Речь становится все более дизартричной, все большее место в ней занимают стереотипные речевые обороты, междометия, вводные слова. Происходят задержки при произнесении начальных букв и слогов, многократное повторение их (логоклоническое заикание). В дальнейшем активная речь ограничивается бессмысленным повторением обрывков слов или отдельных звуков. Почти полностью утрачиваются способности чтения, письма, счета.
Начальная неловкость движений со временем превращается в неспособность к наиболее автоматизированным, жизненно необходимым действиям. Больные как бы разучиваются вставать, садиться, ходить. Они молча лежат, почти не меняя положения.
Продолжительность болезни от 1—2 до 8—10 лет. Смерть чаще наступает в ходе присоединившейся респираторной инфекции.
Болезнь Пика
Заболевание описано А. Пиком в конце XIX века. Оно обычно начинается постепенно в возрасте 40—65 лет. Особенно часто его первые проявления возникают в 55—60 лет.
На начальной стадии болезни Пика в отличие от болезни Альцгеймера преобладают эмоционально-волевые нарушения, а не расстройства интеллектуально-мнестической сферы. Особенно характерна спонтанность, безучастность, пассивность, отсутствие внутренних побуждений к деятельности при сохраняющейся способности к действиям под влиянием стимулов извне. Реже выявляются симптомокомплекс, клинически сходный с картиной прогрессивного паралича, в виде снижения морально-этического уровня личности, беспечности, эйфории, расторможения влечений, некритического отношения к собственному поведению (псевдопаралитический синдром).
Одно из отличий болезни Пика от болезни Альцгеймера заключается в преобладании нарастающей интеллектуальной недостаточности (ослабление способностей к обобщению и абстрагированию, построению адекватных суждений и умозаключений, установлению причинно-следственных зависимостей) над расстройствами памяти.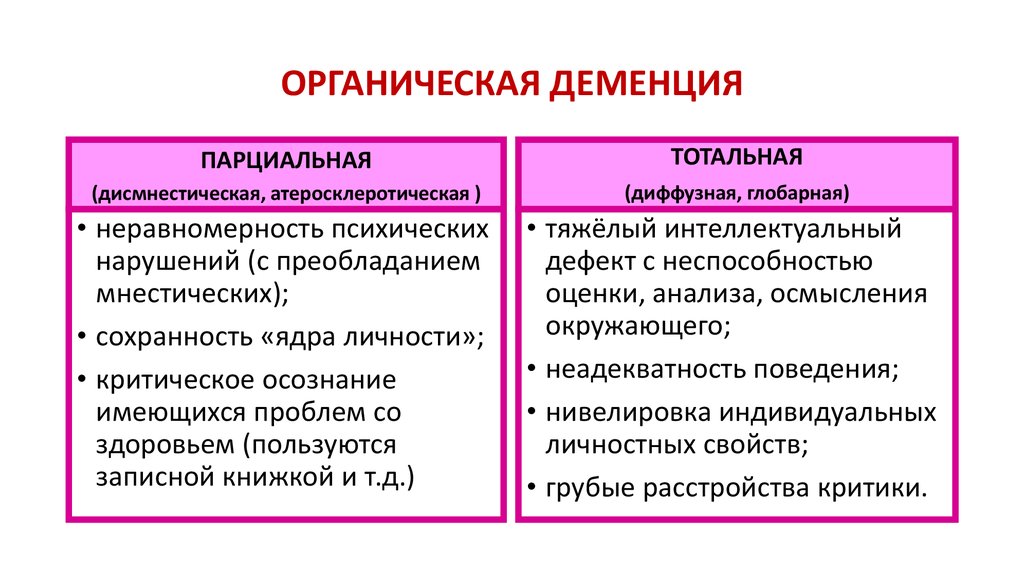 Выраженные нарушения памяти возникают поздно, амнестическая дезориентировка отсутствует. Значительно реже, чем при болезни Альцгеймера, возникают галлюцинаторно-бредовая симптоматика и эпилептиформные припадки.
Выраженные нарушения памяти возникают поздно, амнестическая дезориентировка отсутствует. Значительно реже, чем при болезни Альцгеймера, возникают галлюцинаторно-бредовая симптоматика и эпилептиформные припадки.
При болезни Пика среди проявлений тотального слабоумия ведущее место занимают расстройства речи, тогда как характерные для болезни Альцгеймера нарушения гнозиса и праксиса выражены значительно меньше.
Расстройства речи, начинаясь с затрудненного понимания чужой речи, смыслового и грамматического упрощения, обеднения собственной речи, со временем переходят в речевую беспомощность. Речь насыщается персеверациями, эхолалиями, постепенно утрачивает фразовый характер, сводится к бессмысленному повторению одних и тех же словосочетаний и слов (“стоячий симптом”, типичный именно для болезни Пика). Позднее наступает мутизм.
У части больных развивается маразм. Больные умирают в результате вторичных инфекций спустя 5—6 лет от начала ослабоумливающего церебрально-атрофического процесса.
Этиология, патогенез и патологическая анатомия. В этиологии болезней Альцгеймера и Пика определенное значение придается генетическим факторам. У большинства больных пресенильными деменциями наследственность психопатологически не отягощена. Вместе с тем выявляются семейные случаи болезни Альцгеймера и Пика. Для родителей, братьев и сестер лиц, страдающих болезнями Альцгеймера и Пика, риск возникновения пресенильной деменции выше, чем в общем населении.
При этих формах психической патологии обнаружены разнообразные отклонения в синтезе белков и их функциях на клеточном уровне, установлены нарушения взаимодействия нейротрансмиттерных систем, сниженная концентрация ацетилхолина, катехоламинов и повышенное содержание некоторых микроэлементов в мозговых тканях. С этими биохимическими сдвигами гипотетически связывают атрофию головного мозга, являющуюся анатомической основой болезней Альцгеймера и Пика.
Церебральные патоморфологические изменения при болезни Альцгеймера сходны с таковыми при сенильной деменции. Их наиболее существенная особенность заключается в избирательном, а не диффузном характере церебрально-атрофического процесса, который при болезни Альцгеймера преимущественно локализуется в височных и теменных долях. Избирательность мозговой атрофии сочетается с ее большей выраженностью.
Их наиболее существенная особенность заключается в избирательном, а не диффузном характере церебрально-атрофического процесса, который при болезни Альцгеймера преимущественно локализуется в височных и теменных долях. Избирательность мозговой атрофии сочетается с ее большей выраженностью.
Как и при старческом слабоумии, микроскопически определяется значительное количество сенильных бляшек. Именно для этого заболевания особенно характерны своеобразные патологические изменения в нейрофибриллах (альцгеймеровское перерождение нейрофибрилл).
При болезни Пика, как и при болезни Альцгеймера, атрофия мозга избирательна, но имеет иную локализацию. В атрофический процесс предпочтительно вовлекаются наряду с височными лобные, а не теменные доли.
Микроскопические изменения в мозге существенно отличаются от микроскопической картины болезни Альцгеймера. Старческие бляшки и альцгеймеровские нейрофибриллы, как правило, не обнаруживаются. Определяются атрофия и гибель части корковых нейронов и набухание нервных клеток за счет особых внутриклеточных образований (телец Пика), а также накопление липоидов в клетках паренхимы мозга и глиоцитах.
Диагностика. Распознавание пресенильных деменций основывается на возникновении в предстарческом возрасте прогрессирующего слабоумия тотального типа. Более раннему выявлению типичных для этих заболеваний интеллектуально-мнестических нарушений и расстройств высшей корковой деятельности способствует применение экспериментально-психологических методик. Диагностическое значение имеют пневмоэнцефалография и компьютерная томография, обнаруживающие атрофию головного мозга, внутреннюю гидроцефалию, расширение желудочков мозга. При разграничении болезней Альцгеймера и Пика учитывают их клинические особенности, приведенные выше, и различия в локализации мозгового атрофического процесса, определяемые при помощи пневмоэнцефалографии и компьютерной томографии (атрофия преимущественно теменных и височных областей при болезни Альцгеймера и лобно-височных при болезни Пика).
Распространенность. Заболеваемость и болезненность пресенильными деменциями изучены недостаточно. Есть данные, что риск возникновения пресенильных деменций равен 0,1%. Среди всех госпитализированных в психиатрические стационары лица, страдающие болезнью Альцгеймера, составляют 0,3—0.5%. Болезнь Пика встречается в 2—4 раза реже болезни Альцгеймера. Эти болезни диагностируются значительно реже, чем сенильное слабоумие. Среди лиц с болезнями Альцгеймера и Пика женщин существенно больше, чем мужчин.
Есть данные, что риск возникновения пресенильных деменций равен 0,1%. Среди всех госпитализированных в психиатрические стационары лица, страдающие болезнью Альцгеймера, составляют 0,3—0.5%. Болезнь Пика встречается в 2—4 раза реже болезни Альцгеймера. Эти болезни диагностируются значительно реже, чем сенильное слабоумие. Среди лиц с болезнями Альцгеймера и Пика женщин существенно больше, чем мужчин.
Прогноз пресенильных деменций крайне неблагоприятный вследствие фатального и быстрого распада психической деятельности и наступления смерти в ближайшие несколько лет от начала болезни.
Лечение и профилактика. Лечение болезней Альцгеймера и Пика практически не отличается от терапии сенильной деменций. Методы лечения, способные замедлить или остановить ослабоумливающий процесс, пока не найдены. Проводится терапия сопутствующих соматических заболеваний и возрастных недугов, отдаляющая в части случаев летальный исход. Психотропные средства в малых дозах назначаются при возникновении психоза, более грубых расстройствах поведения и нарушениях сна. Исключительно важен систематический уход за больными.
Исключительно важен систематический уход за больными.
Экспертиза. Больные пресенильными деменциями нетрудоспособны, неспособны к самообслуживанию и недееспособны. В случае совершения правонарушений они признаются невменяемыми.
Пресенильная деменция Определение и значение
- Основные определения
- Тест
- Примеры
/ (priːˈsiːnaɪl) /
Сохранить это слово!
См. синонимы пресенильной деменции на сайте Thesaurus.com
существительное
форма слабоумия неизвестной причины, начинающаяся до того, как человек состарится
Плавно переходите к этим распространенным грамматическим ошибкам, которые ставят многих людей в тупик. Удачи!
Вопрос 1 из 7
Заполните пропуск: Я не могу понять, что _____ подарил мне этот подарок.
Слова, близкие к пресенильному слабоумию
presell, присутствие, камера присутствия, присутствие разума, presenile, presenile dementia, presenility, Present, Presentable, Present Arms, Presentation
Collins English Dictionary — Complete & Unabridged 2012 Digital Edition
© William Collins Sons & Co.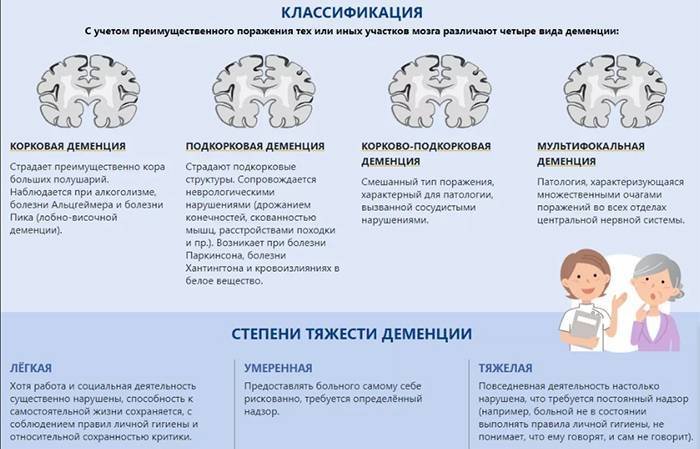 Ltd., 1979, 1986 © HarperCollins
Издательство 1998, 2000, 2003, 2005, 2006, 2007, 2009 гг., 2012
Ltd., 1979, 1986 © HarperCollins
Издательство 1998, 2000, 2003, 2005, 2006, 2007, 2009 гг., 2012
Как использовать пресенильное слабоумие в предложении
Правда, правда, бегемоты благодарности, такие как Sears и Kmart, удваивают ставку на старческое слабоумие.
Walmart снимает проклятие Черной пятницы|Джеймс Пулос|26 ноября 2014 г.|DAILY BEAST
В зависимости от того, какие части мозга поражены, у человека могут развиться формы слабоумия и изменения личности.
Понимание черепно-мозговой травмы Трейси Морган|Джин Ким|20 ноября 2014 г.|DAILY BEAST
Позже могут возникнуть когнитивные и поведенческие проблемы; деменция не редкость.
Бремя, которое нес Робин Уильямс: диагноз: болезнь Паркинсона и депрессия|Dr. Ананд Вееравагу, доктор медицинских наук, Тедж Азад|15 августа 2014 г.|DAILY BEAST
Пожалуй, нет лучшего примера, чем то, как мы относимся к нашим пожилым людям, живущим с деменцией.

Как iPod может бороться с болезнью Альцгеймера и деменцией|Dr. Билл Томас|20 июля 2014 г.|DAILY BEAST

На мой взгляд, это самый вдохновляющий фильм о деменции на сегодняшний день.
Как iPod может бороться с болезнью Альцгеймера и деменцией|Dr. Билл Томас|20 июля 2014 г.|DAILY BEAST
В ряде случаев после исчезновения периода возбуждения отмечается некоторое слабоумие.
Essays In Pastoral Medicine|Austin Malley
Это слабоумие прогрессирует до тех пор, пока, наконец, не наступает состояние почти полного уничтожения умственных способностей.
Essays In Pastoral Medicine|Austin Malley
Если мы не примем его поведение за результат кратковременного слабоумия, вызванного перенапряжением, оно должно оставаться необъяснимым.
Как-то хорошо|Уильям де Морган
Я подозреваю, что все невысказанные в наших душах вещи имеют некоторую вялость бреда и слабоумия.
Тоно Бунгей|Х. G. Wells
Во многих случаях наблюдается стойкое снижение умственных способностей, заканчивающееся деменцией или идиотией.
Британская энциклопедия, 11-е издание, том 9, раздел 6|Разное

ПРЕНАЛЬНОЕ ДЕМЕНЦИЯ И БОЛЕЗНЬ АЛЬЦГЕЙМЕРА ПРИ МОНГОЛИЗМЕ1 | Мозг
Фильтр поиска панели навигации МозгЭтот выпускЖурналы о мозгеНеврологияНеврологияКнигиЖурналыOxford Academic Термин поиска мобильного микросайта
Закрыть
Фильтр поиска панели навигации МозгЭтот выпускЖурналы о мозгеНеврологияНеврологияКнигиЖурналыOxford Academic Термин поиска на микросайте
Расширенный поиск
Журнальная статья
Получить доступ
МЭРИ И. ОЛСОН,
ОЛСОН,
МЭРИ И. ОЛСОН
Ищите другие работы этого автора на:
Оксфордский академический
пабмед
Google ученый
ЧЭН-МЭЙ ШОУ
ЧЕН-МЭЙ ШОУ
Ищите другие работы этого автора на:
Оксфордский академический
пабмед
Google ученый
Мозг , том 92, выпуск 1, март 1969 г., страницы 147–156, https://doi.org/10.1093/brain/92.1.147
Опубликовано:
1 марта 1969 г.
1
1 История статьи
Получено:
18 19 июля68
Опубликовано:
01 марта 1969 г.
- Содержание статьи
- Рисунки и таблицы
- видео
- Аудио
- Дополнительные данные
Цитировать
Cite
MARY I.
OLSON, CHENG-MEI SHAW, PRESENILE DEMENTIA AND ALZHEIMER’S BASE IN MONGOLISM, Brain , Volume 92, Issue 1, March 1969, Pages 147–156, https://doi.org/30.109 /мозг/92.1.147Выберите формат Выберите format.ris (Mendeley, Papers, Zotero).enw (EndNote).bibtex (BibTex).txt (Medlars, RefWorks)
Закрыть
Разрешения
- Электронная почта
- Твиттер
- Фейсбук
- Подробнее
 OLSON, CHENG-MEI SHAW, PRESENILE DEMENTIA AND ALZHEIMER’S BASE IN MONGOLISM, Brain , Volume 92, Issue 1, March 1969, Pages 147–156, https://doi.org/30.109 /мозг/92.1.147
OLSON, CHENG-MEI SHAW, PRESENILE DEMENTIA AND ALZHEIMER’S BASE IN MONGOLISM, Brain , Volume 92, Issue 1, March 1969, Pages 147–156, https://doi.org/30.109 /мозг/92.1.147Фильтр поиска панели навигации МозгЭтот выпускЖурналы о мозгеНеврологияНеврологияКнигиЖурналыOxford Academic Термин поиска мобильного микросайта
Закрыть
Фильтр поиска панели навигации МозгЭтот выпускЖурналы о мозгеНеврологияНеврологияКнигиЖурналыOxford Academic Термин поиска на микросайте
Расширенный поиск
Предварительный просмотр первой страницы статьи PDF
Закрыть
Этот контент доступен только в формате PDF.
© Издательство Оксфордского университета
© Oxford University Press
Раздел выпуска:
Статьи
В настоящее время у вас нет доступа к этой статье.
Скачать все слайды
Войти
Получить помощь с доступом
Получить помощь с доступом
Доступ для учреждений
Доступ к контенту в Oxford Academic часто предоставляется посредством институциональных подписок и покупок. Если вы являетесь членом учреждения с активной учетной записью, вы можете получить доступ к контенту одним из следующих способов:
Доступ на основе IP
Как правило, доступ предоставляется через институциональную сеть к диапазону IP-адресов. Эта аутентификация происходит автоматически, и невозможно выйти из учетной записи с IP-аутентификацией.
Войдите через свое учреждение
Выберите этот вариант, чтобы получить удаленный доступ за пределами вашего учреждения. Технология Shibboleth/Open Athens используется для обеспечения единого входа между веб-сайтом вашего учебного заведения и Oxford Academic.
- Нажмите Войти через свое учреждение.
- Выберите свое учреждение из предоставленного списка, после чего вы перейдете на веб-сайт вашего учреждения для входа в систему.
- Находясь на сайте учреждения, используйте учетные данные, предоставленные вашим учреждением. Не используйте личную учетную запись Oxford Academic.
- После успешного входа вы вернетесь в Oxford Academic.
Если вашего учреждения нет в списке или вы не можете войти на веб-сайт своего учреждения, обратитесь к своему библиотекарю или администратору.
Войти с помощью читательского билета
Введите номер своего читательского билета, чтобы войти в систему. Если вы не можете войти в систему, обратитесь к своему библиотекарю.
Если вы не можете войти в систему, обратитесь к своему библиотекарю.
Члены общества
Доступ члена общества к журналу достигается одним из следующих способов:
Войти через сайт сообщества
Многие общества предлагают единый вход между веб-сайтом общества и Oxford Academic. Если вы видите «Войти через сайт сообщества» на панели входа в журнале:
- Щелкните Войти через сайт сообщества.
- При посещении сайта общества используйте учетные данные, предоставленные этим обществом. Не используйте личную учетную запись Oxford Academic.
- После успешного входа вы вернетесь в Oxford Academic.
Если у вас нет учетной записи сообщества или вы забыли свое имя пользователя или пароль, обратитесь в свое общество.
Вход через личный кабинет
Некоторые общества используют личные аккаунты Oxford Academic для предоставления доступа своим членам. Смотри ниже.
Смотри ниже.
Личный кабинет
Личную учетную запись можно использовать для получения оповещений по электронной почте, сохранения результатов поиска, покупки контента и активации подписок.
Некоторые общества используют личные аккаунты Oxford Academic для предоставления доступа своим членам.
Просмотр учетных записей, вошедших в систему
Щелкните значок учетной записи в правом верхнем углу, чтобы:
- Просмотр вашей личной учетной записи и доступ к функциям управления учетной записью.
- Просмотр институциональных учетных записей, предоставляющих доступ.
Выполнен вход, но нет доступа к содержимому
Oxford Academic предлагает широкий ассортимент продукции. Подписка учреждения может не распространяться на контент, к которому вы пытаетесь получить доступ. Если вы считаете, что у вас должен быть доступ к этому контенту, обратитесь к своему библиотекарю.
Ведение счетов организаций
Для библиотекарей и администраторов ваша личная учетная запись также предоставляет доступ к управлению институциональной учетной записью. Здесь вы найдете параметры для просмотра и активации подписок, управления институциональными настройками и параметрами доступа, доступа к статистике использования и т. д.
Покупка
Стоимость подписки и заказ этого журнала
Варианты покупки книг и журналов в Oxford Academic
Кратковременный доступ
Чтобы приобрести краткосрочный доступ, пожалуйста, войдите в свой личный аккаунт выше.
У вас еще нет личного кабинета? регистр
ПРЕДСЕНИЛЬНАЯ ДЕМЕНЦИЯ И БОЛЕЗНЬ АЛЬЦГЕЙМЕРА В МОНГОЛИЗМЕ 1 — Круглосуточный доступ
ЕВРО €41,00
32 фунта стерлингов
52 доллара США.