Болезнь атрофирование мышц: Врач СПбГПМУ рассказала об опыте лечения спинальной мышечной атрофии
почему в России не лечат взрослых со СМА — Такие дела
- СМА
- Контекст
- Как устроено
- 29. 11. 2021
Сложнейшая ситуация с лекарствами для детей со спинальной мышечной атрофией, кажется, начала разрешаться. Это огромная победа родителей, врачей и благотворительных фондов. Только пока эта победа не для всех: взрослые со СМА все еще выброшены из системы помощи
Это огромная победа родителей, врачей и благотворительных фондов. Только пока эта победа не для всех: взрослые со СМА все еще выброшены из системы помощи
Но СМА не исчезает с возрастом, когда ребенок вырастает
А иногда может даже проявиться и в 20, и в 30 лет — в зависимости от типа заболевания.
Причина болезни одинаковая что у взрослых, что у детей — поломка в гене. И взрослые пациенты нуждаются в такой же поддержке, как и дети.
«Взрослые выкинуты из системы оказания медицинской помощи. Мы говорим о людях с профессией, которые могут работать, которые хотят реализовываться в жизни, заводить семью. Они поставлены в ситуацию “выживай как хочешь, ты можешь рассчитывать только на свои силы и силы своей семьи”», — объясняет Ольга Германенко, директор фонда «Семьи СМА».
«Активный человек, несмотря на возможность управлять только мышкой»
Елене Кузьминой 38 лет, она живет в Муроме — «столице праздника День семьи, любви и верности». Елена работает юристом в фонде «Семьи СМА», путешествует и помогает волонтерскому движению. Любит кино, караоке и играть в бочче — игру, похожую на керлинг.
Любит кино, караоке и играть в бочче — игру, похожую на керлинг.
Мама Лены любя называет дочь «ответственной по связям с общественностью»: к ней приходят с просьбами решить проблемы, позвонить-выяснить, что-то организовать. «Я активный человек, занимаюсь всем», — говорит девушка про себя.
Лена передвигается на инвалидной коляске и почти утратила все двигательные функции. Она может только управлять мышкой и печатать сообщения на виртуальной клавиатуре — именно так мы и договорились об интервью.
Когда Лене был год, родители заметили, что девочка, вместо того чтобы развиваться дальше — ходить, бегать, как будто откатывается назад. Сначала ей поставили диагноз «прогрессирующая мышечная атрофия» и только в 1995 году, когда ученые-генетики открыли ген SMN1, стали писать, что у нее СМА.
«Врачи сказали маме, что такие дети долго не живут — максимум три года, “рожайте другого ребенка”. Поначалу мама действительно ждала, когда я начну умирать. Но прошли три года, пять лет… — тянет Лена. — Мы жили обычной жизнью с поправкой на то, что есть какие-то ограничения».
— Мы жили обычной жизнью с поправкой на то, что есть какие-то ограничения».
«Мы» — это сама Лена и Оля, ее младшая сестра. У девочек разница в возрасте два года, у Оли тоже оказалась СМА.
Инженер-строитель по профессии, Татьяна Алексеевна оставила работу через несколько лет после рождения дочерей, чтобы быть рядом с девочками. «Конечно, для меня это был шок, — вспоминает она. — Никакой поддержки от государства не было, пенсии были копеечные. Муж работал один, финансово было очень тяжело, но сказать, что мы голодали, — нет, такого не было. Я шила одежду и на себя, и на детей. Девчонки обе были умные, учились хорошо. Гостей принимали, праздники отмечали, и мы в гости ездили, на пикники».
В начальных классах мама возила Лену в школу. В семь лет она уже не могла ходить и пересела в коляску, но сама писала в тетрадках, могла перекладывать книжки, играть с игрушками. Позже они с сестрой перешли на домашнее обучение: началась «кабинетная» система и возить на занятия обеих девочек маме стало сложно.
После школы Лена поступила в институт и выучилась на юриста. Писала длинные конспекты на лекциях — тогда она еще могла делать это сама.
В 2009 году Оля умерла от воспаления легких. До этого умер отец девочек — онкологическое заболевание.
Лена с мамой остались вдвоем.
Иллюстрация: Наталья Ямщикова для ТД
Постепенно у Лены уходили навыки — и силы. Сейчас она самостоятельно может только дышать, глотать и разговаривать, одним нажатием отвечать на звонки. Если телефон будет лежать на столе, девушка не сможет поднять его к уху.
«Я никогда самостоятельно не одевалась: футболку еще могла сама на руки натянуть, но чтобы полностью одеться — нет, — говорит Лена. — Никогда не готовила и не помню, чтобы сама переворачивалась в кровати. Всегда будила маму, чтобы она меня перевернула».
Женщины живут вдвоем и стараются справляться со всем сами, «особо никого не напрягая». При необходимости помогают родственники: «По сути, меня утром посадить, покормить, и я могу полдня провести за компьютером — мне больше ничего не нужно, — скромно говорит Лена. — Сходить в туалет, пообедать, и снова занимаюсь делами. Вечером — уложить спать».
— Сходить в туалет, пообедать, и снова занимаюсь делами. Вечером — уложить спать».
«Со стороны, наверное, кажется: “Господи, как они там живут?” — добавляет Татьяна Алексеевна. — Но мы живем и живем. По мере сложностей стараемся совместно приспосабливаться. Это просто наша жизнь. Мы не можем какие-то вещи делать — ну, живем другими делами и событиями».
«Делать шаг назад не хочется»
В августе 2019 года в России зарегистрировали первый препарат для пациентов со СМА. Терапия не излечивает СМА полностью, но останавливает развитие болезни и улучшает качество жизни человека.
Лена сразу же начала собирать документы, но получила лекарство только спустя 20 месяцев — через суд. Ей сделали пять инъекций, шестая будет в феврале 2022 года.
Уже после первых доз Лена почувствовала улучшения. Внешне, может быть, и не видно, но дышать ей стало свободнее и легче, появилось больше энергии и выносливости, нет такой разбитости и вялости. Даже окружающие заметили, что ее голос стал звонче и сильнее.
«Были выборы, бюллетени принесли домой. Я подумала: “Как же расписываться, если мне подпись поставить тяжело?” В итоге взяла ручку и три подписи поставила свободно. Улучшение, получается», — бодро говорит Лена.
Получать лекарство пациенты должны постоянно. Каждый незапланированный перерыв — это риск потерять функцию.
Перестать поднимать руку. Или шевелить пальцами
«Конечно, страшно, если вдруг препарат отменят или перестанут закупать, не хотелось бы чувствовать откат. Уже сейчас, зная, как может быть хорошо, делать шаг назад не хочется. Пускай я не буду ходить без опоры, бегать, но если вернется навык письма ручкой — прекрасно! — убеждена Лена. — Смогу печатать на компьютере свободно — вообще шикарно. Поднимать кружку — это значительная победа».
«Нет категорий “детская” и “взрослая” СМА»
Нельзя сказать, что есть какой-то возрастной порог жизни со СМА: болезнь проявляется в разном возрасте и может развиваться с разной скоростью. Люди со СМА могут жить примерно до 65 лет.
Однако после 25 лет количество взрослых пациентов резко сокращается, говорит Ольга Германенко. Это связано не только с естественным течением болезни, но и с уровнем медицинской помощи взрослым пациентам в России. «Взрослых не хотят лечить, они никому не нужны», — с горечью говорит она.
Взрослых пациентов лечат теми же препаратами, воздействующими на генетический механизм заболевания, что и детей (в России от СМА зарегистрированы препараты рисдиплам и нусинерсен. — Прим. ТД). Но поскольку лекарства от СМА изобрели совсем недавно, к их появлению многие взрослые пациенты уже столкнулись с необратимыми последствиями.
Те, кто утратил большую часть функций, прекрасно понимают, что лекарства не помогут им встать с коляски, но хотя бы вернут выносливость и некоторые навыки: держать ложку, наливать себе чай, писать ручкой, печатать на клавиатуре.
Чем быстрее начнется лечение, тем больше у человека шансов на жизнь. Каждый сбереженный мотонейрон — это сохраненная функция.
Главное для людей со СМА — чтобы их состояние перестало ухудшаться. И опросы, которые проводит фонд среди своих подопечных, подтверждают: все это происходит благодаря лечению.
Только добыть терапию не так-то просто.
Иллюстрация: Наталья Ямщикова для ТД
Если детям со СМА начали выдавать лекарства, в том числе благодаря «Кругу добра», то взрослые остаются с носом и вынуждены бороться за лечение месяцами, иногда дольше. Их должны обеспечивать на региональном уровне, подчеркивает Ольга, но местные власти не горят желанием покупать дорогие лекарства.
Многие пациенты вынуждены судиться. Иногда депздравы выдают лекарства и в досудебном порядке. Бывает и наоборот: даже решение суда не гарантирует, что регион закупит пациенту препарат. Где-то людям выдают лекарство в течение месяца-двух после вынесенного решения — и это еще считается быстро. В иных регионах исполнения решения суда приходится добиваться через судебных приставов.
Проблемы с выдачей лекарств есть в каждом российском регионе, подчеркивает Ольга Германенко.
Нет ни одного субъекта, где лекарства от СМА выдавали бы без проблем и задержек
Самый сложный регион, по оценке фонда «Семьи СМА», — Москва. Из 37 пациентов здесь не получает лечение ни один взрослый. Люди несколько раз выходили на пикеты, Минздрав проводил с ними встречи, но конкретных решений так и не было принято. Примерно в такой же ситуации Свердловская область, «которая детей еще более-менее лечила, а вот со взрослыми просто намертво». Тюменская область «гоняет пациентов по кругу, просто выматывая их».
«Если за детей бегают молодые родители, то за взрослых пациентов бегают уже бабушки или в крайнем случае сами пациенты — люди, многие из которых не могут сами себе налить стакан воды. Все происходит через слезы, через боль и безумную борьбу», — говорит Ольга.
По ее мнению, в ситуации со взрослыми пациентами со СМА снова требуется вмешательство федерального центра, как когда-то было с детьми. Необходимо либо переводить всю нозологию на федеральный бюджет, либо заставлять регионы выполнять законы.
Необходимо либо переводить всю нозологию на федеральный бюджет, либо заставлять регионы выполнять законы.
«Бегу наперегонки с мальчиком и понимаю, что не могу»
С Петром Воротынцевым мы встречаемся в ресторане в центре Москвы. Мужчину сопровождает красивая молодая девушка — его жена Барбора. Петру 33 года, он доцент кафедры истории театра и кино и специалист по истории музыкального театра, преподает на историко-филологическом факультете Российского государственного гуманитарного университета.
У него СМА третьего типа, диагноз ему поставили в детстве. По его воспоминаниям, в детстве он выглядел как здоровый человек, «разве что по лестницам чуть медленнее и хуже ходил». Это не мешало Петру выступать на соревнованиях по настольному теннису за школу.
— Проиграл, правда, в первом туре, — смеется Воротынцев. — Победить мне было важно, потому что я очень амбициозный человек. Ведь без желания победить бессмысленно и заниматься чем-либо.
— Теперь понятно, почему вы с таким упорством боретесь за лекарства, — говорю я.
Петр — один из активных участников московских пикетов, которые устраивают взрослые пациенты со СМА, чтобы добиться положенных им лекарств.
«Спорт — это все-таки честный поединок, — возражает Петр. — А [в ситуации с лекарствами] правила не совсем понятны и не прозрачны. Честно говоря, я бы с радостью не занимался этими пикетами. Я по своей природе вообще не гражданский активист. Мне интереснее книжку почитать».
До 20—22 лет, вспоминает Воротынцев, он еще мог сам в метро ездить, хотя было уже тяжело и, как он осознает сейчас, даже рискованно. Потом стали труднее даваться подъемы по лестницам, затем стало сложно вставать. В 2013 году Петр сломал руку, а в 2018-м — ногу. Теперь он передвигается на коляске, но, если есть возможность хотя бы немного пройти с опорой, ходит до последнего.
«Я стараюсь минус обратить в плюс, — говорит Петр. — Когда я пересел в коляску, то стал больше времени проводить сидя и лежа. По итогу у меня появилось гораздо больше времени, я успеваю писать намного больше текстов».
Иллюстрация: Наталья Ямщикова для ТД
Сейчас Петр в основном работает удаленно, и это тоже плюс. Чтобы сходить в институт на две лекции по полтора часа, ему нужен целый день. Много времени уходит на то, чтобы одеться-раздеться, особенно зимой, и подняться на этаж.
— Бытовые заботы в нашей семье, конечно, по большей части на жене. Если жены нет рядом, то очень помогают родители. Я, что называется, хорошо устроился.
— Он нас поддерживает морально, — твердо говорит Барбора. — Это немало.
Петр и Барбора познакомились в 2009 году в Праге — на родине Барборы, на вокальном мастер-классе. «Петя очень хорошо поет, у него потрясающий голос, даже когда он говорит», — рассказывает Барбора с небольшим акцентом.
В тот год ей самой поставили диагноз «рассеянный склероз», и она ощущала себя «самой больной в мире». После знакомства Барбора и Петр общались на расстоянии, по скайпу, а когда девушка окончила консерваторию, то переехала в Москву. Спустя год у пары родился сын Андрюша.
Спустя год у пары родился сын Андрюша.
Во время разговора Петр берет чайник и наливает себе в чашку чай. Барбора подается вперед, чтобы ему помочь, но, помедлив, отступает.
«Да, налить себе чай я могу, — комментирует Петр. — Но главное, что я могу печатать с iPad и компьютера. Функция правой руки, по крайней мере, сохранена. Левая чуть хуже после перелома, но по сравнению с другими пациентами со СМА мне не на что жаловаться. Этот процесс уже необратим без лекарств, это чистая биология и физиология».
«Пока вопрос не решат с детьми, до нас не дойдет»
Заниматься получением лекарства от СМА Петр начал в конце 2020 года. Пандемия, признается Воротынцев, сильно замедлила этот процесс.
«В 2020 году с детьми еще была большая проблема, — добавляет Барбора. — Всем взрослым, в принципе, было понятно, что, пока вопрос не решат с детьми, до нас не дойдет, что правильно».
Читайте также Обычная девочка на коляске
Сначала Петр обратился в поликлинику — и его «отправили по длинному кругу на госпитализацию». Тогда по совету фонда «Семьи СМА» мужчина пошел в Научный центр неврологии. Пройдя длинный путь обследований, он получил заключение комиссии: ему показана патогенетическая терапия СМА.
Тогда по совету фонда «Семьи СМА» мужчина пошел в Научный центр неврологии. Пройдя длинный путь обследований, он получил заключение комиссии: ему показана патогенетическая терапия СМА.
«И тут-то началось самое интересное», — говорит Воротынцев.
Институт отказался выдавать лекарство без решения региональной комиссии в Центре орфанных заболеваний — его власти Москвы создали через неделю после первого пикета взрослых пациентов.
— С конца июля нас начали настойчиво отправлять туда. В итоге люди выходят оттуда с отказами от врачебной комиссии, — поясняет Петр.
— На основании чего врачебная комиссия отказывает, если есть диагноз и показано лечение?
— Мы задавали этот вопрос на встрече главному внештатному неврологу Москвы, — отвечает Петр. — Он сказал, что отказы выносятся на основании электромиографии: в мышцы колют иголки, чтобы проверить импульсы. Если импульсы плохие, то заключение на лечение они выдавать не будут, это нецелесообразно. Но в этом и суть болезни, что импульсы не проходят!
— Главный невролог фактически проявлением болезни аргументирует отказ в лечении, — подхватывает Барбора. — Естественно, результат будет плохим! Даже если руки или ноги плохо работают, есть еще мышцы сердца, дыхание. Это же тоже нужно поддерживать!
— Естественно, результат будет плохим! Даже если руки или ноги плохо работают, есть еще мышцы сердца, дыхание. Это же тоже нужно поддерживать!
Петр намеренно отказывается от госпитализации, потому что понимает, что получит отказ от врачебной комиссии, как и другие пациенты со СМА. С этим решением добиться положительного решения в суде будет невозможно.
«Чиновники готовы делать все. Они говорят: “Мы вам пандусы организуем, но только не лекарства”. Даже собак предложили выгуливать. Это чем-то похоже на то, как родители детьми не занимаются и пытаются их задобрить, машинку хотя бы купить», — шутит Воротынцев.
Петр обратился в суд. Заседание по его делу прошло 17 ноября. Как Петр и ожидал, ему отказали в лечении, потому что у него не было заключения врачебной комиссии из Центра орфанных заболеваний.
Петр надеется благодаря лечению «хотя бы сохраниться в том состоянии, в котором находится сейчас», и прожить как можно дольше со своей семьей. Закончить научные и творческие проекты.
«Смертность человека — эволюционный двигатель прогресса во всех сферах. Страх тебя подстегивает что-то делать здесь и сейчас, не откладывать жизнь до следующего понедельника. Как рассуждают многие обыватели? Если человек не встанет, не пойдет и не будет, как Жан-Клод Ван Дамм, разбивать кирпичи, то чего его и лечить, собственно. Но если человек проживет на семь — десять лет больше, вы представьте, сколько можно успеть за это время!»
Иллюстрация: Наталья Ямщикова для ТД
«После 18 лет — путь в никуда»
Многие пациенты со СМА сталкиваются с проблемами сразу после совершеннолетия, говорит Ольга Германенко. Так случилось с 18-летним Даниилом Дугушкиным из Москвы.
У парня второй тип СМА. Он успел пролечиться полгода, а потом депздрав перестал выдавать ему лекарства: ребенок стал совершеннолетним.
«За время приема лекарства мое состояние улучшилось. Ощущение, как будто руки сильнее стали, сидеть дольше мог. Сейчас мне страшно, что болезнь начнет прогрессировать. Чувствую, что становится хуже. Меньше выносливости, слабее организм, устаю быстрее. Сидеть могу уже не так долго, как раньше», — говорит Даниил.
Чувствую, что становится хуже. Меньше выносливости, слабее организм, устаю быстрее. Сидеть могу уже не так долго, как раньше», — говорит Даниил.
Он немногословный и очень скромный, даже отказался от видеосвязи. Сухо и обыденно перечисляет свой досуг дома: «Встаю поздно, умываюсь, одеваюсь, выпиваю специальный витаминный коктейль вместо завтрака. Потом работаю — у меня есть подработки на фрилансе — или в интернете сижу. Затем обедаю, читаю книги или слушаю аудиокниги. Иногда выхожу гулять или в торговый центр, в кино. Но сейчас пандемия».
Диагноз Даниилу поставили в 2006 году, рассказывает его мама Елена. В год и два месяца мальчик стал опираться на предметы при ходьбе, но сам переворачивался и садился. С семи лет он передвигается на коляске, но некоторые функции у него сохранились. Например, он может сам есть, чистить зубы, пользоваться телефоном или компьютером, даже играть. Несмотря на это, парню нужна постоянная помощь в быту.
«Днем помогает бабушка, пока я на работе, — рассказывает мама Даниила Елена.
Свое совершеннолетие Даниил встретил в апреле в Морозовской больнице. Там прошел консилиум, который постановил , что парень пожизненно нуждается в терапии. Но поликлиника по месту жительства заключение Морозовской больницы не приняла и направила на госпитализацию повторно.
Суд отказал Дугушкиным в препарате из-за непройденной врачебной комиссии.
«Я так пессимистически к этому отношусь, зная наше государство. Не верю, что нам будут что-то выдавать. У меня страхов нет, я вообще реалист по жизни.
«Это ловушка психики — думать, что вот сейчас я что-то получу — и все будет идеально, — рассуждает Петр о том, что будет, если ему наконец выдадут лекарства. — Поможет — хорошо. Я стараюсь жить ото дня ко дню. А там, я верю, божественное провидение выведет. Но главное — лекарство принадлежит нам по праву. Мне больше даже за ребят обидно. Несправедливость по отношению к другим ранит меня больше. К несправедливости к себе ты, может быть, до определенной степени привык. А несправедливость к другим становится общей».
* * *
Неизвестно, сколько в России живет взрослых людей со СМА. Общего федерального регистра пациентов нет — только регионы ведут учет. В списках фонда «Семьи СМА» зафиксировано 1,1 тысячи пациентов, из них более 300 — старше 18 лет.
Реальное количество людей со СМА, в том числе и взрослых, намного выше.
Статья подготовлена в партнерстве с подразделением фармацевтических товаров «Янссен» ООО «Джонсон & Джонсон»
ООО «Джонсон & Джонсон», Россия, 121614, Москва, улица Крылатская, дом 17, корпус 2.
Контактный телефон: 8 (495) 755-83-57; факс: 8 (495) 755-83-58.
CP-278014
Еще больше важных новостей и хороших текстов от нас и наших коллег — в телеграм-канале «Таких дел». Подписывайтесь!
СМА
Спинальная мышечная атрофия (СМА) — First Genetics
Особенности метода
←
→
Наследственное дегенеративное заболевание, характеризующееся слабостью и истощением (атрофией) скелетных мышц. Слабость и истощение (атрофия) скелетных мышц вызвано снижением количества определенных типов нервных клеток, называемых нижними двигательными нейронами, которые идут от спинного мозга к мышцам и позволяют контролировать их движение.
Слабость, как правило, более выражена в мышцах, расположенных близко к центру тела (проксимальные), по сравнению с мышцами, находящимися далеко от центра тела (дистальные). Начало проявлений мышечной слабости колеблется от пренатального периода до подросткового возраста.
Детская СМА является наиболее распространенной генетической причиной детской смертности 1.
СМА обычно делится на 4 типа в зависимости от возраста начала заболевания и тяжести его течения:
Тип I
Младенческий, болезнь Верднига-Гофмана
Является наиболее распространенной формой этого заболевания. Манифестация заболевания происходит при рождении или в течение первых нескольких месяцев жизни. У детей наблюдаются нарушения моторики/двигательной активности, трудности с дыханием, глотанием, они не могут самостоятельно держать голову и сидеть. Большинство детей со СМА I типа не доживают до двухлетнего возраста из-за дыхательной недостаточности.
Тип II
Промежуточный, болезнь Дубовица
Заболевание обычно проявляется в возрасте от 7 до 18 месяцев. Дети могут сидеть без поддержки, однако не могут стоять или ходить без посторонней помощи. Они часто имеют тремор в пальцах, сколиоз, а также слабость дыхательных мышц, которая может быть опасна для жизни. Продолжительность жизни людей со СМА II типа варьируется от степени вовлечения в патологический процесс дыхательных мышц.
Тип III
Юношеский, болезнь Кюгельберга-Веландер
Манифестация заболевания происходит после 18 месяцев. Пациенты могут стоять и ходить без посторонней помощи, но имеют тенденцию к инвалидизации. Люди со СМА III типа обычно имеют нормальную продолжительность жизни.
Тип IV
Взрослая форма
СМА IV типа встречается редко и начинается в среднем после 35 лет. Пациенты испытывают легкую или умеренную мышечную слабость, тремор и отдышку. Люди со СМА IV типа имеют нормальную продолжительность жизни.
Патогенез
Ключевую роль в развитии СМА играют гены: SMN1 и SMN2, которые содержат инструкции по созданию белка выживания моторных нейронов (Survival of motor neuron protein-interacting protein — SMN). Структура этих генов практически одинакова, они отличаются последовательностью нуклеотидов в 7 и 8 экзонах.
У большинства людей со спинальной мышечной атрофией отсутствует часть гена SMN1, что ухудшает выработку белка SMN. Недостаток белка SMN приводит к гибели моторных нейронов, в результате чего сигналы не передаются от мозга к мышцам. Это приводит к тому, что многие скелетные мышцы становятся слабыми и истощаются, что проявляется в виде нижеописанных симптомов СМА.
Недостаток белка SMN приводит к гибели моторных нейронов, в результате чего сигналы не передаются от мозга к мышцам. Это приводит к тому, что многие скелетные мышцы становятся слабыми и истощаются, что проявляется в виде нижеописанных симптомов СМА.
SMN1 и SMN2гены, играющие ключевую роль в развитии СМА
Количество копий гена
SMN2Как правило, люди имеют две копии гена SMN1 и от одной до двух копий гена SMN2 в каждой клетке. Тем не менее, количество копий гена SMN2 варьируется, причем у некоторых людей встречается до восьми копий. Количество копий гена SMN2 при отсутствии SMN1 изменяет тяжесть состояния и помогает определить тип развивающейся атрофии. Чем больше копий SMN2, тем лучше пациент сможет восполнить потерю SMN1. Для все типов СМА характерно отсутствие части гена SMN1, а число копий SMN2 для каждого типа различно.
Тип I
имеют 1 или 2 копии SMN2
Тип III
3-4 копии SMN2
Тип II
3 копии SMN2
Тип IV
4 и более копий SMN2
Частота
Спинальная мышечная атрофия встречается у 1 из 8000 — 10 000 человек во всем мире.
Тип I
наиболее распространенный тип, на долю которого приходится около половины всех случаев заболевания
Типы II и III
менее распространенные
Тип IV
редкий
Тип наследования
Спинальная мышечная атрофия наследуется по аутосомно-рецессивному типу. В большинстве случаев каждый из родителей человека с аутосомно-рецессивным заболеванием несет по одной копии мутированного гена. Но так как мутация компенсирована наличием второй «здоровой» копией гена, носители не проявляют признаков и симптомов болезни.
Люди, у которых есть более двух копий гена SMN2, обычно не наследуют дополнительные копии от родителя. Возникают они во время случайной ошибки в процессе образования половых клеток (яйцеклетки или сперматозоида) или сразу после оплодотворения.
Эти риски являются приблизительными, поскольку около 2% людей со СМА имеют de novo (возникший спонтанно) вариант SMN1 по одной копии. В этих случаях только один родитель является носителем и для последующей беременности повышенного риска развития СМА нет 2.
Клинические симптомы СМА
Особенности метода
В 2002 году Шоутен и соавторы сообщили о методе мультиплексной амплификации лигированных зондов (MLPA). Этот метод был применен для обнаружения потери или удвоения протяженных участков ДНК.
MLPA
Наиболее чувствительный тест для определения СМА. Он позволяет диагностировать СМА в 95% случаев.
Секвенирование гена
SMN1Для остальных пациентов проводится секвенирование гена SMN1 (он нужен, когда диагноз не удалось подтвердить с помощью MLPA, а у пациента есть клинические проявления СМА). Проводится для остальных 5% пациентов.
В случае, когда ни MPLA, ни секвенирование не помогли найти причину клинических проявлений, которые позволяют врачу заподозрить СМА или другое нервно-мышечное заболевание, врач может назначить исследование панели генов, мутации которых характерны для нервно-мышечных заболеваний.
Биологические образцы, пригодные для данного исследования:
- Периферическая кровь;
- Кровь плода;
- Карты пятен засохшей крови.

Цитируемая литература:
1. Arnold ES, Fischbeck KH. Spinal muscular atrophy. Handb Clin Neurol. 2018;148:591-601
2. Cusin V, Clermont O, Gerard B, Chantereau D, et al. Prevalence of SMN1 deletion and duplication in carrier and normal populations: implication for genetic counseling. J Med Genet 2003; 40
3. Kolb et al. Natural history of infantile-onset spinal muscular atrophy. Ann Neurol. 2017 Dec;82(6):883-891
4. Prior et al. Spinal Muscular Atrophy. GeneReviews®. Adam MP, Ardinger HH, Pagon RA, et al., editors. Seattle (WA): University of Washington, Seattle; 1993-2018
5. Schouten JP, McElguun CJ, Waaijer R, Zwijnenburg D, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002; 30: e57.
Как пройти ДНК тест на СМА?
1
Позвонить 8 (800) 201-74-63 или оставить заявку
2
Оплатить исследование на сайте
Стоимость 8 500 ₽
3
Сдать венозную кровь
Сдавать кровь (4 мл. ) в пробирку с ЭДТА (с фиолетовой крышкой).
) в пробирку с ЭДТА (с фиолетовой крышкой).
Обязательно подпишите каждую пробирку!
4
Сразу позвонить курьеру 8 (800) 201-74-63
Курьер заберет набор с вашей кровью и бесплатно отвезет в лабораторию.
5
Получить результат на эл. почту через 14 рабочих дней
ДНК тест на СМА в лаборатории First Genetics
Специалисты
Многолетний опыт работы в области генетики, лабораторной диагностики и биоинформатики
Конфиденциальность
Все данные строго конфиденциальны и не могут быть переданы третьим лицам
Надежность
Особый контроль на каждом этапе проведения исследования
Срок
Результаты в короткие сроки
Бесплатная доставка
Доставка биоматериала по всей России
Спинальная мышечная атрофия Информационный бюллетень
Что такое спинальная мышечная атрофия?
Что вызывает СМА?
Как наследуется?
Какие существуют типы СМА?
Как диагностируется СМА?
Существуют ли методы лечения СМА?
Каков прогноз?
Какие исследования проводятся?
Где я могу получить дополнительную информацию?
Что такое спинальная мышечная атрофия?
Спинальная мышечная атрофия (СМА) это группа наследственных заболеваний, которые постепенно разрушают моторные нейроны — нервные клетки в стволе головного мозга и спинном мозге, которые контролируют основную активность скелетных мышц, такую как речь, ходьба, дыхание и глотание, приводит к мышечной слабости и атрофии. Моторные нейроны контролируют движения рук, ног, груди, лица, горла и языка. При нарушении передачи сигналов между двигательными нейронами и мышцами мышцы постепенно ослабевают, начинают истощаться и появляются подергивания (называемые 9).0017 фасцикуляции) .
Моторные нейроны контролируют движения рук, ног, груди, лица, горла и языка. При нарушении передачи сигналов между двигательными нейронами и мышцами мышцы постепенно ослабевают, начинают истощаться и появляются подергивания (называемые 9).0017 фасцикуляции) .
top
Что вызывает СМА?
Наиболее распространенная форма СМА вызывается дефектами в обеих копиях гена выживающего двигательного нейрона 1 (SMN1) на хромосоме 5q. Этот ген продуцирует белок двигательных нейронов выживания (SMN), который поддерживает здоровье и нормальную функцию двигательных нейронов. У людей со СМА недостаточный уровень белка SMN, что приводит к потере двигательных нейронов в спинном мозге, вызывая слабость и истощение скелетных мышц. Эта слабость часто более выражена в мышцах туловища и верхней части ног и рук, чем в мышцах кистей и стоп.
Существует много типов спинальной мышечной атрофии, вызванных изменениями в одних и тех же генах. Менее распространенные формы СМА вызываются мутациями в других генах, включая ген VAPB, расположенный на 20-й хромосоме, ген DYNC1h2 на 14-й хромосоме, ген BICD2 на 9-й хромосоме и ген UBA1 на Х-хромосоме. Типы различаются по возрасту начала и выраженности мышечной слабости; однако между типами есть совпадение.
Типы различаются по возрасту начала и выраженности мышечной слабости; однако между типами есть совпадение.
top
Как наследуется?
За исключением редких случаев, вызванных мутациями в гене UBA1, СМА наследуется в0017 аутосомно-рецессивный , что означает, что у пораженного человека есть два мутировавших гена, часто наследуемых по одному от каждого родителя. Те, у кого только один мутировавший ген, являются носителями болезни без каких-либо симптомов. Аутосомно-рецессивные заболевания могут поражать более одного человека в одном поколении (братьев, сестер или двоюродных братьев).
top
Какие существуют типы SMA?
Существует широкий спектр нарушений, наблюдаемых при СМА, вызванных дефектами гена SMN1, от начала до рождения с затрудненным дыханием при рождении до легкой слабости у взрослых. Соответственно, эту наиболее распространенную форму СМА можно разделить на четыре типа в зависимости от достигнутого наивысшего моторного уровня.
- СМА I типа, также называемая болезнью Верднига-Гоффмана или СМА с инфантильным началом , обычно проявляется в возрасте до 6 месяцев. Наиболее тяжело пораженные дети (СМА типа 0 или IA) имеют ограниченные движения даже внутриутробно и рождаются с контрактурами и затрудненным дыханием, при этом смерть наступает в первый год жизни без лечения. Симптомы СМА I типа включают гипотонию (снижение мышечного тонуса), снижение подвижности конечностей, отсутствие сухожильных рефлексов, фасцикуляции, затруднения при глотании и кормлении, а также нарушение дыхания. У этих детей также развивается сколиоз (искривление позвоночника) или другие скелетные аномалии по мере взросления. Без какого-либо лечения пораженные дети никогда не сидят и не стоят, и подавляющее большинство обычно умирает от дыхательной недостаточности в возрасте до 2 лет. Дети со СМА типа I теперь живут дольше и могут достигать более высоких двигательных навыков, таких как сидение и даже ходьба, благодаря более активной клинической помощи и новому доступному лечению, модифицирующему заболевание.
- Дети с СМА типа II, промежуточной формой , обычно проявляют свои первые симптомы в возрасте от 6 до 18 месяцев, хотя у некоторых они могут проявиться раньше. Они могут сидеть без поддержки, но не могут стоять или ходить без посторонней помощи, а некоторые могут со временем потерять способность самостоятельно сидеть без лечения. У них могут быть проблемы с дыханием, включая гиповентиляцию во сне. Прогрессирование заболевания вариабельно без лечения. Ожидаемая продолжительность жизни сокращается, но большинство людей доживает до подросткового или юношеского возраста. Благодаря лечению, модифицирующему заболевание, и активной клинической помощи дети со СМА II типа улучшили двигательные исходы.
- У детей с СМА типа III (болезнь Кугельберга-Веландера) симптомы развиваются после 18 месяцев, и они могут ходить самостоятельно. Сначала они испытывают трудности при ходьбе и беге, подъеме по ступенькам или вставании со стула. Чаще всего первыми поражаются проксимальные мышцы ног, при этом наблюдается тремор рук. Осложнения включают сколиоз и контрактуры суставов — хроническое укорочение мышц или сухожилий вокруг суставов, вызванное аномальным мышечным тонусом и слабостью, что препятствует свободному движению суставов. Люди со СМА типа III могут быть склонны к респираторным инфекциям, но при осторожном обращении большинство из них имеют нормальную продолжительность жизни. Лечение, модифицирующее заболевание, может улучшить результаты.
- У лиц со СМА типа IV симптомы развиваются после 21 года, с легкой или умеренной слабостью проксимальных мышц и другими симптомами.

 Осложнения включают сколиоз и контрактуры суставов — хроническое укорочение мышц или сухожилий вокруг суставов, вызванное аномальным мышечным тонусом и слабостью, что препятствует свободному движению суставов. Люди со СМА типа III могут быть склонны к респираторным инфекциям, но при осторожном обращении большинство из них имеют нормальную продолжительность жизни. Лечение, модифицирующее заболевание, может улучшить результаты.
Осложнения включают сколиоз и контрактуры суставов — хроническое укорочение мышц или сухожилий вокруг суставов, вызванное аномальным мышечным тонусом и слабостью, что препятствует свободному движению суставов. Люди со СМА типа III могут быть склонны к респираторным инфекциям, но при осторожном обращении большинство из них имеют нормальную продолжительность жизни. Лечение, модифицирующее заболевание, может улучшить результаты.top
Как диагностируется СМА?
Доступен анализ крови для поиска делеций или мутаций гена SMN1. Этот тест выявляет не менее 95 процентов СМА типов I, II и III, а также может выявить, является ли человек носителем дефектного гена, который может быть передан детям. Если ген SMN1 не является аномальным или анамнез и результаты обследования не типичны для СМА, другие диагностические тесты могут включать электромиографию (которая регистрирует электрическую активность мышц во время сокращения и в состоянии покоя), исследование скорости нервной проводимости (которое измеряют способность нерва посылать электрический сигнал), биопсию мышц (используется для диагностики многих нервно-мышечных расстройств) и другие анализы крови.
top
Существуют ли методы лечения СМА?
Полного излечения от СМА не существует. Лечение заключается в управлении симптомами и предотвращении осложнений.
В декабре 2016 года Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США одобрило нусинерсен (Спинраза™) в качестве первого препарата, одобренного для лечения детей и взрослых со СМА. Препарат вводят путем инъекции в жидкость, окружающую спинной мозг. Он предназначен для увеличения производства полноразмерного белка SMN, который имеет решающее значение для поддержания двигательных нейронов. Польза лучше всего задокументирована у младенцев и детей, особенно при раннем начале. Несколько других методов лечения находятся на поздних стадиях разработки и могут стать доступными для пострадавших людей в ближайшем будущем.
В мае 2019 г. Управление по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) одобрило генную терапию онасемногеном абепаровек-ксиои (Zolgensma™) для детей в возрасте до 2 лет с инфантильной СМА. Безопасный вирус доставляет полностью функциональный ген SMN человека к целевым двигательным нейронам, что, в свою очередь, улучшает движение и функцию мышц, а также повышает выживаемость. В августе 2020 года FDA одобрило пероральный препарат рисдиплам (Еврисди) для лечения пациентов в возрасте двух месяцев и старше со СМА.
Безопасный вирус доставляет полностью функциональный ген SMN человека к целевым двигательным нейронам, что, в свою очередь, улучшает движение и функцию мышц, а также повышает выживаемость. В августе 2020 года FDA одобрило пероральный препарат рисдиплам (Еврисди) для лечения пациентов в возрасте двух месяцев и старше со СМА.
Физиотерапия, трудотерапия и реабилитация могут помочь улучшить осанку, предотвратить неподвижность суставов и замедлить мышечную слабость и атрофию. Упражнения на растяжку и укрепление могут помочь уменьшить контрактуры, увеличить диапазон движений и поддерживать нормальное кровообращение. Некоторым людям требуется дополнительная терапия для устранения проблем с речью и глотанием. Вспомогательные устройства, такие как опоры или скобы, ортопедические стельки, синтезаторы речи и инвалидные коляски, могут помочь улучшить функциональную независимость.
Правильное питание и калории необходимы для поддержания веса и силы при избегании длительного голодания. Людям, которые не могут жевать или глотать, может потребоваться введение зонда для кормления. Неинвазивная вентиляция ночью может улучшить дыхание во время сна, а некоторым людям также может потребоваться вспомогательная вентиляция днем из-за слабости мышц шеи, горла и грудной клетки.
Людям, которые не могут жевать или глотать, может потребоваться введение зонда для кормления. Неинвазивная вентиляция ночью может улучшить дыхание во время сна, а некоторым людям также может потребоваться вспомогательная вентиляция днем из-за слабости мышц шеи, горла и грудной клетки.
наверх
Каков прогноз?
Прогноз зависит от типа СМА. Некоторые формы СМА без лечения приводят к летальному исходу.
Люди со СМА могут казаться стабильными в течение длительного времени, но без лечения нельзя ожидать улучшения.
top
Какие исследования проводятся?
Национальный институт неврологических расстройств и инсульта (NINDS), входящий в состав Национальных институтов здравоохранения (NIH), проводит фундаментальные, трансляционные и клинические исследования СМА в лабораториях NIH, а также поддерживает исследования посредством грантов для крупных медицинских учреждений. учреждений по всей стране.
Клеточные и молекулярные исследования направлены на понимание механизмов, вызывающих дегенерацию двигательных нейронов.
Ученые проанализировали ткани человека и разработали широкий спектр модельных систем на животных и клетках для изучения болезненных процессов и ускорения тестирования потенциальных методов лечения. Среди этих усилий:
- Было показано, что генная терапия и специальные лекарства останавливают разрушение двигательных нейронов и замедляют прогрессирование заболевания на моделях мышей и у людей со СМА. NINDS поддерживает исследования, направленные на разработку этих методов и создание условий для клинических испытаний на пациентах. Клинические испытания генной терапии при СМА продолжаются.
- Модели СМА на животных представляют собой важные инструменты для открытия и разработки новых методов лечения СМА. Ученые разработали модели рыбок данио, мышей и свиней, в том числе модели менее тяжелых типов СМА 2 и 3, которые могут значительно помочь в определении новых терапевтических мишеней и потенциальных методов лечения.
Ученые, поддерживаемые NIH, собрали продольные данные о досимптомных или недавно диагностированных детях со СМА типов 1, 2 или 3 и их здоровых братьях и сестрах.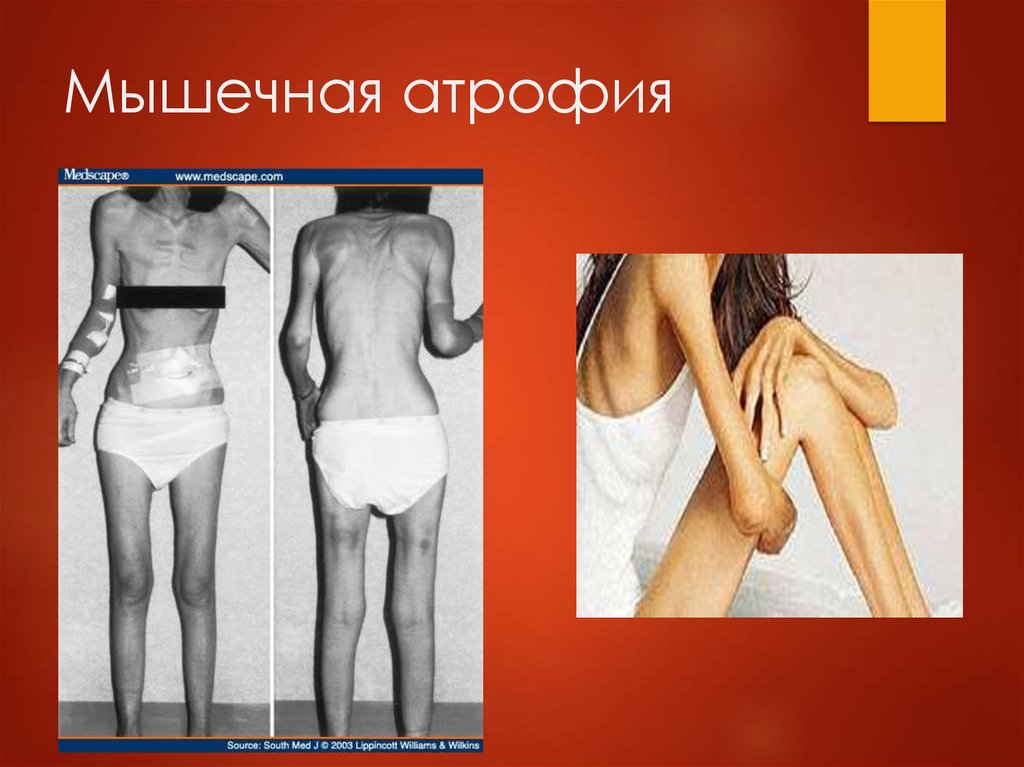 Целью данного исследования является консультирование и информирование родителей о возможных возможностях проведения клинических испытаний.
Целью данного исследования является консультирование и информирование родителей о возможных возможностях проведения клинических испытаний.
NINDS создала сеть клинических испытаний NeuroNext (NINDS Network for Excellence in Neuroscience Clinical Trials) для содействия быстрой разработке и проведению испытаний неврологических расстройств, поражающих взрослых и/или детей. Среди своих целей сеть предназначена для разработки испытаний на ранней стадии, направленных на выявление биомаркеров — обычно физических признаков или веществ в крови или других жидкостях организма, которые можно измерить для определения наличия и тяжести заболевания — и тестирования перспективных, появляющиеся терапии. Один из проектов заключался в том, чтобы определить биомаркеры СМА и понять причину и механизмы, лежащие в основе заболевания. Данные естественной истории, полученные в ходе этого исследования, привели к решению об одобрении нусинерсена. Знания, полученные в ходе этого исследования, позволили улучшить дизайн дополнительных клинических испытаний СМА.
Где я могу получить дополнительную информацию?
Для получения дополнительной информации о неврологических расстройствах или исследовательских программах, финансируемых Национальным институтом неврологических расстройств и инсульта, обращайтесь в Сеть ресурсов и информации о мозге (BRAIN) Института по телефону:
BRAIN
P.O. Box 5801
Bethesda, MD 20824
800-352-9424
Информация также доступна в следующих организациях:
Cure SMA
925 Busse Road
Elk Grove Village, IL 60007
847-367-7620
800-886-1762
Фонд мускулистого атрофии спинала
126 Восточная 56-я улица
30-й этаж
Нью-Йорк, Нью-Йорк 10022
646-253-71003
New York, NY 10022
646-253-71003.
877-386-3762
Ассоциация мышечной дистрофии
161 N. Clarke,
Suite 3550
Чикаго, IL 60601
800-572-1717
«Spinal Muscular Atrophy.
Публикация NIH № 19-NS-5597
Back to Spinal Muscular Atrophy Information Page
See a list of all NINDS disorders
Publicaciones en Español
Atrofia Muscular Espinal
Prepared by:
Office of Communications and Public Liaison
National Institute of Neurological Disorders and Stroke
National Institutes of Health
Bethesda, MD 20892
Медицинские материалы NINDS предоставляются только в информационных целях и не обязательно отражают одобрение или официальную позицию Национального института неврологических расстройств и Инсульт или любое другое федеральное агентство. Рекомендации по лечению или уходу за отдельным пациентом должны быть получены путем консультации с врачом, который осматривал этого пациента или знаком с историей болезни этого пациента.
Вся информация, подготовленная NINDS, находится в открытом доступе и может быть свободно скопирована. Приветствуется кредит NINDS или NIH.
Приветствуется кредит NINDS или NIH.
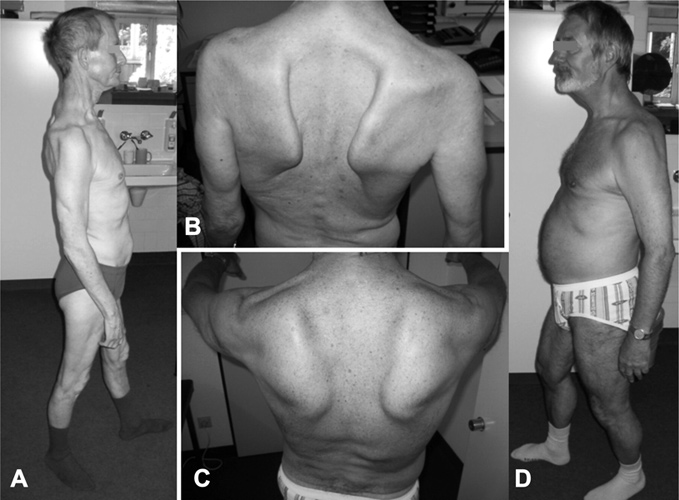
Мышечная атрофия: причины, симптомы и диагностика
Мышечная атрофия
Мышечная атрофия — это истощение мышц. Обычно это связано с недостатком физической активности.
Когда из-за болезни или травмы вам трудно или невозможно двигать рукой или ногой, отсутствие подвижности может привести к атрофии мышц. Со временем, без регулярных движений, ваша рука или нога могут начать казаться меньше, но не короче, чем та, которую вы можете двигать.
В некоторых случаях атрофию мышц можно остановить с помощью правильной диеты, физических упражнений или физиотерапии.
У вас может быть атрофия мышц, если:
- Одна из ваших рук или ног заметно меньше другой.
- Вы испытываете заметную слабость в одной конечности.
- Вы были физически неактивны в течение очень долгого времени.
Позвоните своему врачу, чтобы назначить полное медицинское обследование, если вы считаете, что у вас может быть атрофия мышц или если вы не можете нормально двигаться. У вас может быть недиагностированное заболевание, требующее лечения.
У вас может быть недиагностированное заболевание, требующее лечения.
Неиспользуемые мышцы могут ослабнуть, если вы не будете активны. Но даже после того, как она началась, этот тип атрофии часто можно обратить вспять с помощью упражнений и улучшения питания.
Мышечная атрофия также может произойти, если вы прикованы к постели или не можете двигать определенными частями тела из-за состояния здоровья. У космонавтов, например, после нескольких дней пребывания в невесомости может произойти атрофия мышц.
Другие причины мышечной атрофии включают:
- отсутствие физической активности в течение длительного периода времени
- старение
- связанная с алкоголем миопатия, боль и слабость в мышцах из-за чрезмерного употребления алкоголя в течение длительного периода времени
- ожоги
- травмы, такие как разрыв вращательной манжеты плеча или переломы костей
- недоедание
- спинной или периферический мозг травмы нерва
- инсульт
- длительная терапия кортикостероидами
Некоторые заболевания могут привести к истощению мышц или могут затруднить движение, что приведет к атрофии мышц. К ним относятся:
К ним относятся:
- боковой амиотрофический склероз (БАС) , также известный как болезнь Лу Герига, поражает нервные клетки, контролирующие произвольное движение мышц
- дерматомиозит , вызывает мышечную слабость и кожную сыпь состояние, которое приводит к воспалению нервов и мышечной слабости
- рассеянный склероз , аутоиммунное состояние, при котором организм разрушает защитную оболочку нервов
- мышечная дистрофия , наследственное заболевание, вызывающее мышечную слабость
- невропатия , повреждение нерва или группы нервов, приводящее к потере чувствительности или функции
- остеоартрит , вызывающий снижение подвижности в суставах полиомиелит , вирусное заболевание, поражающее мышечную ткань, которое может привести к параличу
- полимиозит , воспалительное заболевание
- ревматоидный артрит , хроническое воспалительное аутоиммунное заболевание, поражающее суставы
- спинальная мышечная атрофия , наследственное заболевание, вызывающее истощение мышц рук и ног
Если мышечная атрофия вызвана другим заболеванием, вам может потребоваться пройти обследование для диагностики условие.
Ваш врач запросит полную историю болезни. Скорее всего, вас попросят:
- сообщить им о старых или недавних травмах и ранее диагностированных заболеваниях
- перечислите рецептурные, безрецептурные лекарства и добавки, которые вы принимаете
- дайте подробное описание ваших симптомов
Ваш врач может также назначить анализы, чтобы помочь в диагностике и исключить определенные заболевания. Эти тесты могут включать:
- анализы крови
- рентгенографию
- магнитно-резонансную томографию (МРТ)
- компьютерную томографию (КТ)
- исследования нервной проводимости
- биопсию мышц или нервов
- Электромиография (ЭМГ)
Ваш врач может направить вас к специалисту в зависимости от результатов этих тестов.
Лечение будет зависеть от вашего диагноза и степени потери мышечной массы. Любые основные медицинские условия должны быть рассмотрены. Общие методы лечения мышечной атрофии включают:
- физические упражнения
- физиотерапию
- ультразвуковую терапию
- хирургическое вмешательство
- диетические изменения
Рекомендуемые упражнения могут включать упражнения в воде, чтобы облегчить движение.