Деменция наследственность: Генетическая предрасположенность к развитию деменции
Генетическая предрасположенность к развитию деменции
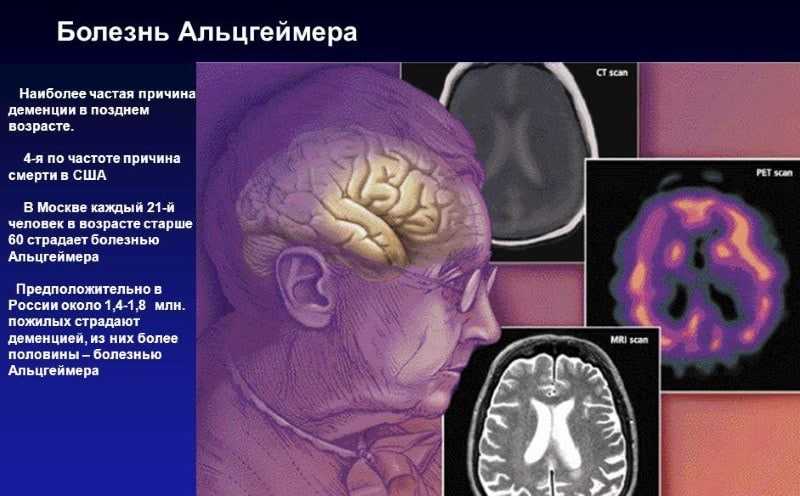
25% населения мира в возрастной группе от 55 лет и старше имеют семейную историю деменции. В большинстве случаев, семейные случаи данной патологии возникают вследствие наличия множества генетических дефектов, которые в совокупности значительно повышают риск развития деменции. Небольшая доля семей среди общей популяции имеет моногенные формы деменции с ранней манифестацией заболевания, которые обусловлены мутацией одного из генов деменции.
В этом обзоре авторы сфокусировали внимание на результатах и методах генетической диагностики болезни Альцгеймера (APP, PSEN1 и PSEN2 гены) с рассмотрением практических аспектов медико-генетического консультирования.
Введение
Когда у кого-то из родственников диагностируется деменция, клиницист часто может слышать вопрос: «У моей мамы деменция. Возможно, у меня тоже разовьется; могу ли я это проверить и предотвратить?»
Надо отметить, что отягченный анамнез по развитию деменции у родственников – необязательно означает наличие моногенной формы заболевания.
Как же идентифицировать то небольшое количество семей с повышенным риском развития деменции?
Менделевская форма заболевания и мультифакториальные заболевания
Каждая семья испытывает на себе влияние как окружающей среды, так и генетических факторов, поэтому семейные заболевания не всегда носят наследственный характер. Наиболее ярким примером деменции, вызванной факторами окружающей среды, является куру. Эта патология является инфекционной прионной болезнью, впервые зарегистрированной в 1950 году на папуасских островах Новой Гвинеи, где родственники употребляли в пищу умершего в погребальных ритуалах. Сначала все считали, что это заболевание генетической природы по причине его семейного характера, но результаты экспериментальных работ показали, что это, по сути, трансмиссивная спонгиформная энцефалопатия.
Однако, в подавляющем большинстве, известные нам факторы риска развития деменции генетической природы.
Генетические факторы могут вносить вклад в развитие семейной деменции двумя путями: приводя к менделевским формам заболевания (моногенным) или в качестве способствующего фактора мультифакториальных (полигенных) болезней (Рис.1).
Рисунок 1. Сравнение патогенеза моногенных и полигенных (мультифакториальных) заболеваний.
Клинические последствия менделевских форм деменции
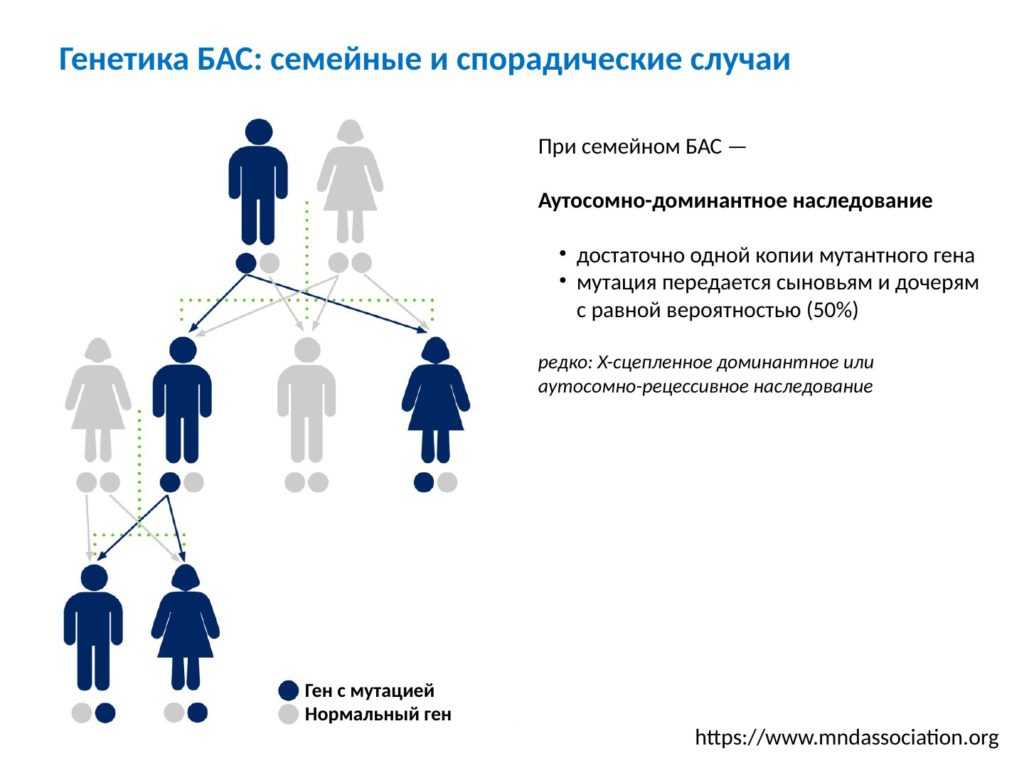
Моногенные формы деменции с установленными генами-виновниками подчиняются аутосомно-доминантным принципам наследования и обладают высокой пенетрантностью, когда наблюдается высокий уровень заболеваемости в последующих поколениях. В этом случае очень полезным оказывается генетическое консультирование. Несмотря на наличие в генеалогическом древе лиц – носителей мутации, но не проявивших признаки заболевания в настоящее время, у этих людей в 95% и выше случаев развивается деменция.
Принципы генетических исследований при деменции
Обдумывание необходимости генетического исследования включает в себя два шага. Первый шаг – детальный анализ истории семьи (наличие заболевания у родственников в нескольких поколениях) с дифференцировкой между моногенным типом наследования и мультифакториальными формами болезни. В данном аспекте семьям с аутосомно-доминантным типом наследования заболевания рекомендуется проведение генетического анализа. Второй шаг на пути к правильному решению – детальный анализ фенотипа заболевания с целью проведения адекватного потребностям генетического теста. К примеру, история психических нарушений как интегральный показатель фенотипа болезни, характерна для фронтотемпоральной деменции.
Генетический аспект болезни Альцгеймера
Клинически, типичная форма болезни Альцгеймера характеризуется градуальным началом и быстрым прогрессированием в виде расстройств памяти и когнитивной дисфункции (по критериям 1984г. Тhe National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer’s Disease and Related Disorders Association [NINCDS-ADRDA]). Эти диагностические критерии были пересмотрены с целью выявления форм болезни без нарушений памяти (с языковыми, зрительно-пространственными дисфункциями) и определения роли биомаркеров в диагностике (критерии 2011г. Тhe National Institute on Aging–Alzheimer’s Association [NIA-AA]).%20%D0%B4%D0%B5%D0%BC%D0%B5%D0%BD%D1%86%D0%B8%D1%8F.jpg)
Моногенные формы болезни Альцгеймера крайне редки: свыше 35 миллионов людей в мире страдают данной патологией, но генетические мутации установлены в случае 500 семей. Ключевыми элементами в анамнезе заболевания, помогающими отдифференцировать менделевское наследование от полигенного, являются: наследование болезни через поколения и ранний возраст манифестации болезни (таблица 1). Члены семей, отвечающие этим параметрам, несут в себе патогенетическую мутацию в одном из установленных для болезни генов. К примеру, Raux c коллегами, изучая когорту из 65 семей с ранней манифестацией заболевания (младше 60 лет) и наличием болезни в трех поколениях выявили: в 86% случаев – мутацию в гене-виновнике заболевания (78% — в виде изменения последовательности нуклеотидов, 8% — в виде патологической дупликации гена). Однако, семьи, отвечающие этим критериям, очень редко встречаются: распространенность в возрастной группе 41-60 лет около 5 на 100 000 человек.
Таблица 1.
| Возраст манифестации симптомов | Вероятность наличия мутации | |
|---|---|---|
| Наличие болезни в трех генерациях | 86% | |
| Наличие болезни у 2-ух и более родственников 1-ой линии | 68% | |
| Наличие болезни у 2-ух и более родственников 1-ой линии | 15% | |
| Наличие болезни у 2-ух и более родственников 1-ой линии | старше 65 лет |
Эта группа семей с аутосомно-доминантным наследованием патологии при манифестации болезни имеет ту же клиническую картину, что и другие формы болезни, однако развитие миоклонуса на ранних стадиях заболевания может быть диагностическим маркером моногенной болезни.
Известны три причинных гена болезни Альцгеймера: APP, PSEN1 и PSEN2. Надо отметить, что первый из них – ген APP на 21 хромосоме был обнаружен в 1987 году у лиц с трисомией 21 (синдромом Дауна), у которых развивается деменция со схожей гистопатологической картиной. Однако ни одной семьи с АРР-мутацией не было зарегистрировано до 1991 года. К тому времени было установлено, что главными гистопатологическими признаками болезни Альцгеймера является наличие амилоидных бляшек и нейрофибриллярных клубков. Дупликация АРР приводит к формированию бета-амилоида – главного компонента амилоидных бляшек. Эта находка легла в основу амилоидной гипотезы, предполагающей, что продукция и деградация бета-амилоида являются основными в патогенезе не только менделевской формы заболевания, но и болезни мультифакториальной, развившейся в общей популяции.
Надо отметить, что первый из них – ген APP на 21 хромосоме был обнаружен в 1987 году у лиц с трисомией 21 (синдромом Дауна), у которых развивается деменция со схожей гистопатологической картиной. Однако ни одной семьи с АРР-мутацией не было зарегистрировано до 1991 года. К тому времени было установлено, что главными гистопатологическими признаками болезни Альцгеймера является наличие амилоидных бляшек и нейрофибриллярных клубков. Дупликация АРР приводит к формированию бета-амилоида – главного компонента амилоидных бляшек. Эта находка легла в основу амилоидной гипотезы, предполагающей, что продукция и деградация бета-амилоида являются основными в патогенезе не только менделевской формы заболевания, но и болезни мультифакториальной, развившейся в общей популяции.
Впоследствии были выявлены еще два дополнительных гена-виновника: PSEN1 на хромосоме 14 и PSEN2 на хромосоме 1. В обоих случаях, мутации приводили к гиперпродукции бета-амилоида или, в некоторых случаях, к изменению соотношения изоформ бета-амилоида1-42 к бета-амилоиду1-40 (рис. 1).
1).
Рисунок 1. Амилоидная гипотеза и патогенез болезни Альцгеймера.
Так сформировалась амилоидная гипотеза, являющаяся доминирующей парадигмой в изучении данного заболевания. Хотя патогенез заболевания в общей популяции, вероятно, более сложный и многофакторный.
Как отмечалось ранее, 86% семей с ранней манифестацией деменции (в возрасте ранее 60 лет) в трех и более поколениях имеют мутации в генах APP, PSEN1 или PSEN2. Мутации PSEN1 – наиболее частая причина, встречающаяся у 60% семей с менделевской формой болезни. Около 15% семей имеют мутацию в виде изменения нуклеотидной последовательности АРР, а в 8% случаев встречается дупликация АРР. Мутации PSEN2 достаточно редка: установлена лишь у 22 семей.
Таким образом, при необходимости, в первую очередь проводится скрининг на наличие мутации PSEN1. Существуют также клинические ключи к выбору приоритетного генетического анализа. Так, наличие болезни Альцгеймера и спастического парапареза более характерно при наличии PSEN1 мутации. В то время как АРР мутация часто приводит к церебральной амилоидной ангиопатии с развитием церебральных кровоизлияний.
В то время как АРР мутация часто приводит к церебральной амилоидной ангиопатии с развитием церебральных кровоизлияний.
Практические аспекты медико-генетического консультирования
Генетическое консультирование обычно проводится у лиц с уже развившимся заболеванием и в целях прогноза. В обоих случаях эта процедура бывает полезной. Разумеется, генетическое исследование у лиц с развившейся болезнью, не влияет на клиническое ведение пациента, но может служить хорошим помощником в постановке диагноза, когда он не столь очевиден, а также, в случае выявления моногенной болезни, быть поводом для консультации всех членов семьи.
Важно, что необходима очная консультация генетика с выяснение всех «точек над и», когда дело касается проведения генетических анализов для определения прогноза развития болезни в будущем. И главное в этом аспекте – понимание, что никаких превентивных мер лечения этой патологии не существует. В таких случаях, члены семьи желают провести генетическое обследование по трем причинам: вопрос функции памяти, планирование будущей жизни и планирование деторождения. Отмечено, что в случае получения положительных результатов существует повышенный риск суицидов, поэтому не рекомендуется проводить обследование у пациентов с психологическими и психиатрическими проблемами.
Отмечено, что в случае получения положительных результатов существует повышенный риск суицидов, поэтому не рекомендуется проводить обследование у пациентов с психологическими и психиатрическими проблемами.
Заключение
К счастью, моногенные формы деменции редко встречаются. Родственники пациентов с развившейся болезнью, имеют риск развития деменции в течение всей жизни около 20% по сравнению с 10% в общей популяции. Однако, небольшая когорта семей с аутосомно-доминантными формами наследования имеют мутацию одного из генов-виновников. Каждый ребенок пациента с моногенным заболеванием имеет 50% шанс иметь ту же мутацию и при наличии её – подвергается 95% риску развития болезни в течение жизни. Около 50% родственников в таких семьях после детальной консультации с генетиком и лечащим врачом отказываются от генетического обследования на предмет выявления генов-виновников еще не развившегося заболевания.
Источник: Clement T Loy, Prof Peter R Schofield, Anne M Turner. Genetics of dementia. The Lancet. Published online August 6, 2013 http://dx.doi.org/10.1016/S0140-6736(13)60630-3
Genetics of dementia. The Lancet. Published online August 6, 2013 http://dx.doi.org/10.1016/S0140-6736(13)60630-3
Наследственные деменции — Редкий журнал
Введение.
Деменция является большой проблемой общественного здравоохранения из-за старения общества. Тем не менее, высокую распространенность деменции в пожилом возрасте может затмить значение её возникновения у более молодых пациентов. Деменции молодого возраста очень трудно диагностируются, но они дают понимание общих представлений о деменции у пожилых пациентов. Например, высокая распространенность наследственных деменций у более молодых возрастных групп привели к идентификации причинных генов и последующей молекулярной патологии, имеющих непосредственное отношение к более общим спорадическим случаям заболевания у пожилых пациентов. Перспектива будущего лечения, направленного на конкретные патологические молекулярные изменения при различных деменция, делает точный диагноз очень важным.
Определения.
Молодые деменции обычно включают пациентов до 65-летнего возраста. Эта возрастная группа является социальным показателем с точки зрения работоспособности и пенсионного возраста, но этот возраст не имеет конкретного биологического значения и существует целый ряд заболеваний, для которых не актуально данное деление.
Терминология деменции в настоящее время включает две особые проблемы. Во-первых, стандартные критерии деменции требуют, что когнитивные нарушения должны быть достаточно серьезными. Во-вторых, должна быть специфически нарушена память. Следствием первой проблемы является задержка в диагностике конкретной причины деменции. Становится все более важно осуществлять специфическую раннюю диагностику причин когнитивных нарушений, особенно учитывая возможность модулирующего лечения в будущем. В недавно предложенных критериях для болезни Альцгеймера важность ранней диагностики и роль биомаркеров, независимо от степени тяжести, являются одинаково признанными.
Вторая проблема заключается в том, что большинство критериев деменции требуют ухудшения именно эпизодической памяти. Это существует потому, что болезнь Альцгеймера является наиболее частой причиной деменции у пожилых и наиболее изучена. Следует заметить, что пациенты с прогрессирующим снижением когнитивных функций без нарушения памяти (например, семантическая деменция) могут быть исключены из диагностического алгоритма деменции. Таким образом, следует придавать особое значение анализу когнитивных и неврологических синдромов у любого пациента со снижением когнитивных функций, и это особенно касается деменций с ранним началом, для которых дифференциальный диагноз часто широк.
Это существует потому, что болезнь Альцгеймера является наиболее частой причиной деменции у пожилых и наиболее изучена. Следует заметить, что пациенты с прогрессирующим снижением когнитивных функций без нарушения памяти (например, семантическая деменция) могут быть исключены из диагностического алгоритма деменции. Таким образом, следует придавать особое значение анализу когнитивных и неврологических синдромов у любого пациента со снижением когнитивных функций, и это особенно касается деменций с ранним началом, для которых дифференциальный диагноз часто широк.
Эпидемиология.
Есть несколько популяционных исследований по эпидемиологии молодых деменций. Харви и коллеги рассчитали, что в двух районах Лондона в Великобритании распространенность деменции с началом в возрасте от 30 до 65 лет составила 54 на 100 000 и 98 на 100 000 в возрасте от 45 до 65 лет. Болезнь Альцгеймера является наиболее распространенной деменцией, далее идут сосудистые заболевания и фронтотемпоральная деменция. Используя двухступенчатый почтовый опрос, Икеджима и коллеги нашли в целом аналогичную распространенность 42 на 100 000 в префектуре Ибараки в Японии в возрасте от 18-65 лет. В отличие от исследований Великобритании, наиболее частой причиной в Японии являются сосудистые заболевания, а затем болезнь Альцгеймера.
Используя двухступенчатый почтовый опрос, Икеджима и коллеги нашли в целом аналогичную распространенность 42 на 100 000 в префектуре Ибараки в Японии в возрасте от 18-65 лет. В отличие от исследований Великобритании, наиболее частой причиной в Японии являются сосудистые заболевания, а затем болезнь Альцгеймера.
Основные причины деменции.
Болезнь Альцгеймера, сосудистые заболевания, фронтотемпоральная деменция и деменция с тельцами Леви являются наиболее распространенными заболеваниями, которые вызывают деменцию как у пожилых, так и у более молодых пациентов, кроме тех, кто моложе 35 лет. Тем не менее, клинические признаки этих заболеваний у более молодых пациентов могут отличаться от тех, у кого болезнь началась в более позднем возрасте.
Болезнь Альцгеймера.
Первому пациенту Альцгеймера было только 51 год на момент обращения и, в течение ближайших 50 лет, болезнь Альцгеймера называли пресенильной деменцией. По сравнению с пожилыми лицами, молодые пациенты имеют меньше сопутствующих заболеваний, таких как заболевания почек или сердечно-сосудистой системы, и более низкое потребление лекарственных препаратов, которые могут усугублять когнитивные нарушения. Аутосомно-доминантная семейная форма болезни Альцгеймера чаще встречается у лиц с ранним началом деменции, спорадические же случаи болезни Альцгеймера у лиц моложе 50 лет встречаются редко. В отличие от пациентов со спорадическими случаями заболевания, у пациентов с семейной болезнью Альцгеймера, как правило, есть миоклонусы, в некоторых случаях, заметные речевые нарушения. У пациентов с пресенилин-1 делециями и некоторыми точечными мутациями спастический парапарез может быть до начала когнитивных нарушений, редко бывает мозжечковая атаксия. ApoEε4 генотип может способствовать более агрессивным клиническим вариантам течения заболевания у молодых пациентов.
Аутосомно-доминантная семейная форма болезни Альцгеймера чаще встречается у лиц с ранним началом деменции, спорадические же случаи болезни Альцгеймера у лиц моложе 50 лет встречаются редко. В отличие от пациентов со спорадическими случаями заболевания, у пациентов с семейной болезнью Альцгеймера, как правило, есть миоклонусы, в некоторых случаях, заметные речевые нарушения. У пациентов с пресенилин-1 делециями и некоторыми точечными мутациями спастический парапарез может быть до начала когнитивных нарушений, редко бывает мозжечковая атаксия. ApoEε4 генотип может способствовать более агрессивным клиническим вариантам течения заболевания у молодых пациентов.
Сосудистая деменция и сосудистые когнитивные нарушения.
У молодых пациентов, могут быть такие редкие причины, как митохондриальная болезнь или церебральная аутосомно-доминантная артериопатия с подкорковыми инфарктами и лейкоэнцефалопатией (CADASIL).
Фронтотемпоральная деменция.
Большая часть приходится на молодых пациентов, что может быть связано с высокой наследуемостью фронтотемпоральной деменции: около 20-40% семейные случаи.
Деменция с тельцами Леви и деменция при болезни Паркинсона.
Деменция с тельцами Леви в молодом возрасте развивается редко. Трипликации α-синуклеина и мутации в гене глюкоцереброзидазы могут быть связаны со значительными когнитивными нарушениями, в ряде случаев напоминающими классическую деменцию с тельцами Леви.
Синдром деменции плюс.
Клиническая концепция синдрома деменции плюс (например, синдромы деменции, в которых когнитивные нарушения сопровождается дополнительными неврологическими или системными нарушениями) могут быть полезны для клинической диагностики и проведения дифдиагноза.
Атаксия.
Целиакия, спиноцеребеллярная атаксия (особенно типа 2, 12 и 17), паранеопластические заболевания, прионные заболевания, синдром Х-сцепленной атаксии и тремора, семейные британские и датские деменции, митохондриальные болезни, поверхностный сидероз, лизосомные болезни накопления центральной нервной системы, болезнь Нимана-Пика типа C, множественные системные атрофии (деменция обычно незначительна, если она присутствует), болезнь Александера и рассеянный склероз.
Пирамидные знаки.
Рассеянный склероз, целиакия, фронтотемпоральная деменция с поражением двигательных нейронов, болезнь Альцгеймера (некоторые пресенильные мутации), спиноцеребеллярные атаксии, фенилкетонурия, семейные британские и датские деменции, наследственный спастический парапарез 4 типа, адренолейкодистрофия.
Дистония и хорея.
Болезнь Гентингтона (и гентингтоноподобные синдромы 1-3), целиакия, болезнь Куфа, болезнь Вильсона, нейроакантоцитоз, пантотенаткиназно-связанные нейродегенерации (нейродегенерации с накоплением железа в ЦНС), синдром Леша-Нихана, кортикобазальные дегенерации, нейроферритинопатии, анти-NMDA-рецептор-опосредованный лимбический энцефалит, болезнь Крейтцфельда-Якоба.
Поражение щек и языка.
Нейроакантоцитоз, синдром Леша-Нихана.
Акинетико-ригидный синдром.
Болезнь с тельцами Леви (деменция с тельцами Леви и деменция при болезни Паркинсона), прогрессирующий надъядерный паралич, множественные системные атрофии (деменция обычно незначительна, если она присутствует), болезнь Гентингтона (особенно при раннем начале заболевания), кортикобазальная дегенерация, хроническая травматическая энцефалопатия, болезнь Вильсона, пантотенаткиназно-связанные нейродегенерации, фронтотемпоральная деменция с паркинсонизмом, болезнь Альцгеймера.
Полиневропатия.
Целиакия, нейроакантоцитоз, церебрально-сухожильный ксантоматоз, ВИЧ-инфекции, аксональные невропатии, алкоголь-связанные заболевания, метахроматическая лейкодистрофия, порфирия, адренолейкодистрофия, GM2-ганглиозидоз, болезнь Краббе, сиалидоз, болезнь Фабри, митохондриальные болезни, спиноцеребеллярные атаксии (в частности, тип 3).
Миоклонус или ранние припадки.
Целиакия, прионные заболевания, болезнь Альцгеймера, болезнь с тельцами Леви, митохондриальные болезни, болезь Гоше, GM2-ганглиозидоз, подострый склерозирующий панэнцефалит, синдром прогрессирующей миоклонической эпилепсии, болезнь Куфа, сиалидоз.
Глазодвигательные нарушения.
Болезнь Нимана-Пика типа С (вертикальный надъядерный паралич взора), болезнь Гоше (горизонтальный надъядерный паралич взора), прогрессирующий надъядерный паралич, митохондриальные болезни, спиноцеребеллярные атаксии (в частности, тип 2), паранеопластические расстройства, болезнь Уиппла.
Глухота.
Поверхностный сидероз, митохондриальные болезни, семейные датские деменции, альфа-маннозидоз, сиалидоз.
Вегетативные нарушения.
Болезнь с тельцами Леви, множественные системные атрофии, прионные заболевания (фатальная семейная инсомния), порфирия, адренолейкодистрофия, анти-NMDA- рецептор-опосредованный лимбический энцефалит.
Желудочно-кишечные дисфункции.
Целиакия, болезнь Уиппла, порфирия. При целиакии идет выработка антител к тканевой трансглютаминазе класса 2 (тТГ). Этот фермент широко распространен в тканях позвоночных (головной мозг, тонкая кишка) и играет критическую роль в контроле клеточного и тканевого гомеостаза, апоптозе. Вследствии поражения тТГ антителами у больных с целиакией могут отмечаться как умеренный когнитивный синдром, так и деменция.
Катаракта.
Миотоническая дистрофия, церебральносухожильный ксантоматоз, митохондриальные болезни, семейные датские деменции.
Спленомегалия.
Болезнь Нимана-Пика типа C, болезнь Гоше.
Сухожильныеь ксантомы.
Церебральносухожильный ксантоматоз.
Болезнь Педжета.
Валозин-связанная фронтотемпоральная деменция.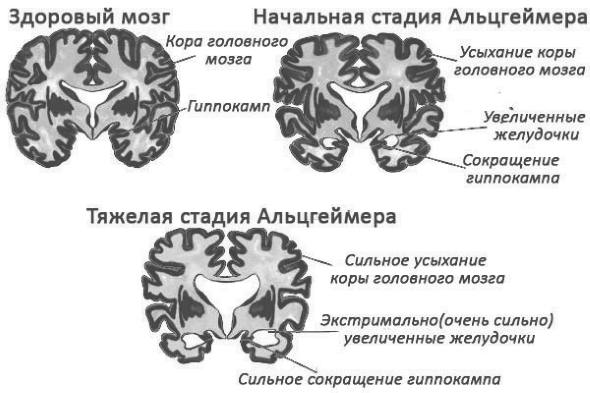
Почечная недостаточность.
Болезнь Фабри, синдром Леша-Нихана, митохондриальные болезни.
Печеночная дисфункция.
Болезнь Вильсона, болезнь Гоше, митохондриальные болезни.
Нарушение дыхания.
Фронтотемпоральная деменция с поражением двигательных нейронов, митохондриальные болезни, анти-NMDA-рецептор-опосредованный лимбический энцефалит.
Анемия.
Дефицит витамина B12, целиакия, нейроакантоцитоз, болезнь Вильсона, болезнь Гоше.
Поражение кожи.
Болезнь Бехчета, целиакия, системные васкулиты и заболевания соединительной ткани, болезнь Фабри.
Метаболические или инфекционные кризы.
Болезнь Александера, целиакия, дефицит орнитинтранскарбамилазы, альфаманнозидоз, порфирия.
Выводы.
Дифференциальная диагностика деменций молодого возраста затруднена, поэтому необходим структурированный клинический подход на основе всех клинических проявлений. Молодые деменции отличаются от деменций пожилого возраста.
текст: С.В.Копишинская, В.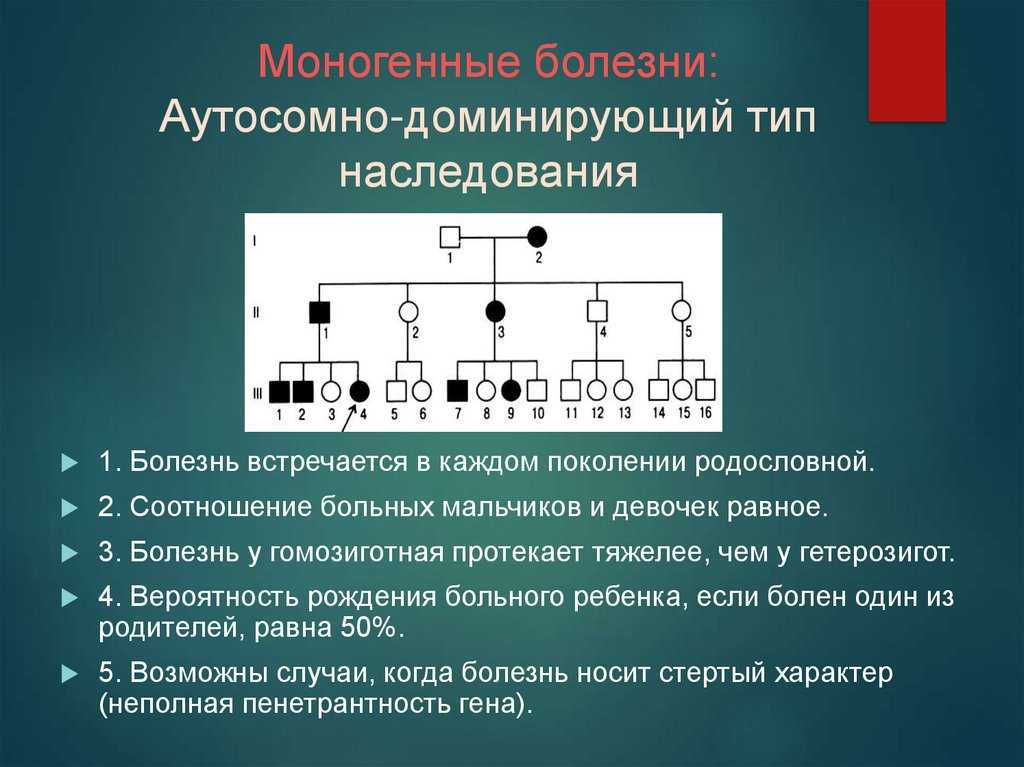 А.Антонова, А.А.Репин, С.Н.Светозарский
А.Антонова, А.А.Репин, С.Н.Светозарский
Кафедра неврологии, психиатрии и наркологии ФПКВ Нижегородской государственной медицинской академии, Нижегородский областной клинический диагностический центр
* * *
Список использованной литературы.
1. Левин О.С. Алгоритмы диагностики и лечения деменции. – Москва, «МЕДпресс-информ». – 2011.
2. Руководство по гериатрической психиатрии // Под ред. С.И. Гавриловой. – Москва, издательство «Пульс». – 2011.
3. Di Sabatino A., Vanoli A., Giuffrida P., Luinetti O. et all. The function of tissue transglutaminase in celiac disease // Autoimmun Rev. – 2012. – Feb. – P.456-463.
4. Rossor M.N., Fox N.C., Mummery C.J., Schott J.M. The diagnosis of young-onset dementia // Lancet Neurol. – 2010. — Aug; 9 (8). – P. 793-806.
Генетика деменции — PubMed
Обзор
. 1 марта 2014 г .; 383 (9919): 828-40.
doi: 10.1016/S0140-6736(13)60630-3. Epub 2013 6 августа.
Epub 2013 6 августа.
Клемент Т Лой 1 , Питер Р. Шофилд 2 , Энн М Тернер 3 , Джон Б. Дж. Квок 4
Принадлежности
- 1 Школа общественного здравоохранения Сиднейского университета, Сидней, Новый Южный Уэльс, Австралия; Neuroscience Research Australia, Рандвик, Новый Южный Уэльс, Австралия; Служба болезней Хантингтона, больница Вестмид, Вестмид, Новый Южный Уэльс, Австралия.
- 2 Neuroscience Research Australia, Рандвик, Новый Южный Уэльс, Австралия; Университет Нового Южного Уэльса, Кенсингтон, Новый Южный Уэльс, Австралия.
- 3 Отделение медицинской генетики Сиднейской детской больницы, Рандвик, Новый Южный Уэльс, Австралия.

- 4 Neuroscience Research Australia, Рандвик, Новый Южный Уэльс, Австралия; Университет Нового Южного Уэльса, Кенсингтон, Новый Южный Уэльс, Австралия. Электронный адрес: [email protected].
- PMID: 23927914
- DOI: 10.1016/С0140-6736(13)60630-3
Обзор
Clement T Loy et al. Ланцет. .
. 1 марта 2014 г .; 383 (9919): 828-40.
doi: 10.1016/S0140-6736(13)60630-3. Epub 2013 6 августа.
Авторы
Клемент Т Лой 1 , Питер Р. Шофилд 2 , Энн М Тернер 3 , Джон Б. Дж. Квок 4
Шофилд 2 , Энн М Тернер 3 , Джон Б. Дж. Квок 4
Принадлежности
- 1 Школа общественного здравоохранения Сиднейского университета, Сидней, Новый Южный Уэльс, Австралия; Neuroscience Research Australia, Рандвик, Новый Южный Уэльс, Австралия; Служба болезней Хантингтона, больница Вестмид, Вестмид, Новый Южный Уэльс, Австралия.
- 2 Neuroscience Research Australia, Рандвик, Новый Южный Уэльс, Австралия; Университет Нового Южного Уэльса, Кенсингтон, Новый Южный Уэльс, Австралия.
- 3 Отделение медицинской генетики Сиднейской детской больницы, Рандвик, Новый Южный Уэльс, Австралия.
- 4 Neuroscience Research Australia, Рандвик, Новый Южный Уэльс, Австралия; Университет Нового Южного Уэльса, Кенсингтон, Новый Южный Уэльс, Австралия. Электронный адрес: [email protected].
 Электронный адрес:
Электронный адрес: - PMID: 23927914
- DOI: 10.1016/С0140-6736(13)60630-3
Абстрактный
25% всех людей в возрасте 55 лет и старше имеют семейную историю деменции. У большинства семейная история связана с генетически сложным заболеванием, при котором многие генетические вариации с небольшим эффектом взаимодействуют, увеличивая риск деменции. Риск развития деменции в течение жизни для этих семей составляет около 20% по сравнению с 10% среди населения в целом. Небольшая часть семей имеет аутосомно-доминантный семейный анамнез ранней деменции, которая часто связана с менделевской болезнью, вызванной мутацией в одном из генов деменции. Каждый член семьи имеет 50-процентный шанс унаследовать мутацию, что дает пожизненный риск деменции более 9 баллов. 5%. В этом обзоре мы сосредоточимся на доказательствах и подходах к генетическому тестированию болезни Альцгеймера (гены APP, PSEN1 и PSEN2), лобно-височной деменции (MAPT, GRN, C9ORF72 и другие гены) и других семейных деменциях. В заключение мы обсудим практические аспекты генетического консультирования.
5%. В этом обзоре мы сосредоточимся на доказательствах и подходах к генетическому тестированию болезни Альцгеймера (гены APP, PSEN1 и PSEN2), лобно-височной деменции (MAPT, GRN, C9ORF72 и другие гены) и других семейных деменциях. В заключение мы обсудим практические аспекты генетического консультирования.
Copyright © 2014 Elsevier Ltd. Все права защищены.
Похожие статьи
Генетическое тестирование семейной AD и FTD: спектр мутаций и фенотипов в датской когорте.
Линдквист С.Г., Шварц М., Батбайли М., Вальдемар Г., Нильсен Дж.Е. Линдквист С.Г. и соавт. Клин Жене. 2009 авг; 76 (2): 205-9. doi: 10.1111/j.1399-0004.2009.01191.x. Epub 2009 29 июля. Клин Жене. 2009. PMID: 19659892
Изучение роли редкой вариабельности кодирования в генах менделевской деменции (APP, PSEN1, PSEN2, GRN, MAPT и PRNP) при болезни Альцгеймера с поздним началом.
Сасси С., Геррейро Р., Гиббс Р., Дин Дж., Луптон М.К., Троукс С., Аль-Саррадж С., Ниблок М., Галло Дж. М., Аднан Дж., Киллик Р., Браун К.С., Медуэй С., Лорд Дж., Тертон Дж., Брас Дж. ; Британский консорциум исследований болезни Альцгеймера, Морган К., Пауэлл Дж. Ф., Синглтон А., Харди Дж. Сасси С. и др. Нейробиол Старение. 2014 дек;35(12):2881.e1-2881.e6. doi: 10.1016/j.neurobiolaging.2014.06.002. Epub 2014 16 июня. Нейробиол Старение. 2014. PMID: 25104557 Бесплатная статья ЧВК.
Генетическое консультирование и тестирование семей с болезнью Альцгеймера.
Ковальская А. Ковальская А. Нейрол Нейрохир Пол. 2004 г., ноябрь-декабрь; 38(6):495-501. Нейрол Нейрохир Пол. 2004. PMID: 15654674 Обзор. польский.
Мутационный анализ семейной деменции с ранним началом у населения Японии.
Роль мутаций PSEN1 и MAPT R406W.Икеучи Т., Канеко Х., Мияшита А., Нодзаки Х., Касуга К., Цукиэ Т., Цучия М., Имамура Т., Исидзу Х., Аоки К., Исикава А., Онодера О., Кувано Р., Нисидзава М. Икеучи Т. и др. Дементное гериатрическое когнитивное расстройство. 2008;26(1):43-9. дои: 10.1159/000141483. Epub 2008 28 июня. Дементное гериатрическое когнитивное расстройство. 2008. PMID: 18587238
Генетическое тестирование и консультирование в диагностике и лечении деменции в молодом возрасте.
Голдман Дж.С. Голдман Дж.С. Психиатр Clin North Am. 2015 июнь;38(2):295-308. doi: 10.1016/j.psc.2015.01.008. Epub 2015 18 марта. Психиатр Clin North Am. 2015. PMID: 25998117 Обзор.

 Роль мутаций PSEN1 и MAPT R406W.
Роль мутаций PSEN1 и MAPT R406W.Посмотреть все похожие статьи
Цитируется
Связь амилоидной, тау- и митохондриальной гипотез болезни Альцгеймера и определение перспективных мишеней для лекарств.
Фишар З. Фишар З. Биомолекулы. 2022 11 ноября; 12 (11): 1676. doi: 10.3390/biom12111676. Биомолекулы. 2022. PMID: 36421690 Бесплатная статья ЧВК. Обзор.
Клинические характеристики и анализ корреляции генотип-фенотип у пациентов с семейной болезнью Альцгеймера с патогенными/вероятно патогенными мутациями предшественников амилоидных белков.
Лю И, Сяо Х, Лю Х, Ляо Х, Чжоу И, Вэн Л, Чжоу Л, Лю Х, Би СЮ, Сюй Т, Чжу И, Ян Ц, Чжан С, Хао Х, Чжан В, Ван Дж, Цзяо Б., Шен Л. Лю Ю и др. Front Aging Neurosci. 2022 14 окт;14:1013295. doi: 10.3389/fnagi.2022.1013295. Электронная коллекция 2022. Front Aging Neurosci. 2022. PMID: 36313020 Бесплатная статья ЧВК.
Предварительная оценка нутрицевтического потенциала ягоды асаи ( Euterpe sp.
) как потенциального природного средства для лечения болезни Альцгеймера.А.Л.Нассер М.Н., Меллор И.Р., Картер В.Г. А.Л.Нассер М.Н. и соавт. Молекулы. 2022 30 июля; 27 (15): 4891. дои: 10.3390/молекулы 27154891. Молекулы. 2022. PMID: 35956841 Бесплатная статья ЧВК.
Активация реакции гипоксии защищает мышей от накопления β-амилоида.
Оллонен Т., Куркела М., Лаитакари А., Сакко С., Койвисто Х., Мюллихарью Дж., Танила Х., Серпи Р., Койвунен П. Оллонен Т. и др. Cell Mol Life Sci. 2022 19 июля; 79 (8): 432. doi: 10.1007/s00018-022-04460-6. Cell Mol Life Sci. 2022. PMID: 35852609Бесплатная статья ЧВК.
ZNF384: потенциальная терапевтическая мишень для псориаза и болезни Альцгеймера через воспаление и метаболизм.
Лю С., Юань С., Су Х., Лю Ф., Чжуан З., Чен Ю. Лю С. и др. Фронт Иммунол. 2022 20 мая; 13:892368. doi: 10.3389/fimmu.2022.892368. Электронная коллекция 2022. Фронт Иммунол. 2022. PMID: 35669784 Бесплатная статья ЧВК.

 ) как потенциального природного средства для лечения болезни Альцгеймера.
) как потенциального природного средства для лечения болезни Альцгеймера.
Просмотреть все статьи «Цитируется по»
Типы публикаций
термины MeSH
Генетика деменции — PMC
1. Bertram L, Lill CM, Tanzi RE. Генетика болезни Альцгеймера: назад в будущее. Нейрон. 2010;68(2):270–281. [PubMed] [Google Scholar]
2. Ван Броекховен К. Будущее генетических исследований нейродегенерации. Нат Мед. 2010;16(11):1215–1217. [PubMed] [Академия Google]
3. Crews L, Masliah E. Молекулярные механизмы нейродегенерации при болезни Альцгеймера. Хум Мол Жене. 2010;19(R1):R12–R20. [Бесплатная статья PMC] [PubMed] [Google Scholar]
4. Kim J, Basak JM, Holtzman DM. Роль аполипопротеина Е в развитии болезни Альцгеймера. Нейрон. 2009;63(3):287–303. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Kim J, Basak JM, Holtzman DM. Роль аполипопротеина Е в развитии болезни Альцгеймера. Нейрон. 2009;63(3):287–303. [Бесплатная статья PMC] [PubMed] [Google Scholar]
5. Green RC, Roberts JS, Cupples LA, et al. Исследовательская группа REVEAL Раскрытие генотипа APOE для риска болезни Альцгеймера. N Engl J Med. 2009 г.;361(3):245–254. [Бесплатная статья PMC] [PubMed] [Google Scholar]
6. Ashe KH, Zahs KR. Изучение биологии болезни Альцгеймера у мышей. Нейрон. 2010;66(5):631–645. [Бесплатная статья PMC] [PubMed] [Google Scholar]
7. Kim D, Tsai LH. Сближение физиологии и патологии при БА. Клетка. 2009;137(6):997–1000. [PubMed] [Google Scholar]
8. Thinakaran G, Koo EH. Транспортировка, процессинг и функция белка-предшественника амилоида. Дж. Биол. Хим. 2008;283(44):29615–29619. [Бесплатная статья PMC] [PubMed] [Google Scholar]
9. ЛаФерла FM. Пути, связывающие Абета- и тау-патологии. Биохим Сок Транс. 2010;38(4):993–995. [PubMed] [Google Scholar]
10. Ittner LM, Ke YD, Delerue F, et al. Дендритная функция тау опосредует токсичность бета-амилоида на моделях мышей с болезнью Альцгеймера. Клетка. 2010;142(3):387–397. [PubMed] [Google Scholar]
Ittner LM, Ke YD, Delerue F, et al. Дендритная функция тау опосредует токсичность бета-амилоида на моделях мышей с болезнью Альцгеймера. Клетка. 2010;142(3):387–397. [PubMed] [Google Scholar]
11. Cruchaga C, Kauwe JS, Mayo K, et al. SNP, связанные с уровнями фосфотау в спинномозговой жидкости, влияют на скорость снижения заболеваемости болезнью Альцгеймера. Генетика PLoS. 2010;6(9):ii, e100110. [Бесплатная статья PMC] [PubMed] [Google Scholar]
12. Fagan AM, Head D, Shah AR, et al. Снижение уровня Abeta(42) в спинномозговой жидкости коррелирует с атрофией головного мозга у когнитивно нормальных пожилых людей. Энн Нейрол. 2009;65(2):176–183. [Бесплатная статья PMC] [PubMed] [Google Scholar]
13. Holtzman DM. Спинномозговая жидкость бета-амилоид 42, тау и Р-тау: подтверждение теперь реализация. Арх Нейрол. 2009;66(12):1552–1553. [PubMed] [Google Scholar]
14. Hu WT, Chen-Plotkin A, Arnold SE, et al. Новые биомаркеры ЦСЖ для болезни Альцгеймера и легких когнитивных нарушений. Акта Нейропатол. 2010;119(6): 669–678. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Акта Нейропатол. 2010;119(6): 669–678. [Бесплатная статья PMC] [PubMed] [Google Scholar]
15. Jack CR, Jr, Knopman DS, Jagust WJ, et al. Гипотетическая модель динамических биомаркеров патологического каскада болезни Альцгеймера. Ланцет Нейрол. 2010;9(1):119–128. [Бесплатная статья PMC] [PubMed] [Google Scholar]
16. Джагуст В.Дж., Ландау С.М., Шоу Л.М. и др. Инициатива по нейровизуализации болезни Альцгеймера Взаимосвязи между биомаркерами старения и деменции. Неврология. 2009;73(15):1193–1199. [Бесплатная статья PMC] [PubMed] [Google Scholar]
17. Morris JC, Roe CM, Grant EA, et al. Питтсбургская визуализация соединения B и прогнозирование прогрессирования от когнитивной нормальности до симптоматической болезни Альцгеймера. Арх Нейрол. 2009;66(12):1469–1475. [Бесплатная статья PMC] [PubMed] [Google Scholar]
18. Вемури П., Висте Х.Дж., Вейганд С.Д. и др. Инициатива по нейровизуализации болезни Альцгеймера Серийные биомаркеры МРТ и ЦСЖ при нормальном старении, MCI и AD. Неврология. 2010;75(2):143–151. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Неврология. 2010;75(2):143–151. [Бесплатная статья PMC] [PubMed] [Google Scholar]
19. Сперлинг Р.А., Айсен П.С., Беккет Л.А. и соавт. На пути к определению доклинических стадий болезни Альцгеймера: рекомендации рабочих групп Национального института старения и Ассоциации Альцгеймера по диагностическим рекомендациям по болезни Альцгеймера. Демент Альцгеймера. 2011;7(3):280–292. [Бесплатная статья PMC] [PubMed] [Google Scholar]
20. Bertram L, McQueen MB, Mullin K, Blacker D, Tanzi RE. Систематический метаанализ исследований генетической ассоциации болезни Альцгеймера: база данных AlzGene. Нат Жене. 2007;39(1): 17–23. [PubMed] [Google Scholar]
21. Colhoun HM, McKeigue PM, Davey Smith G. Проблемы сообщения о генетических ассоциациях со сложными результатами. Ланцет. 2003;361(9360):865–872. [PubMed] [Google Scholar]
22. Гарольд Д., Абрахам Р., Холлингворт П. и др. Полногеномное ассоциативное исследование выявляет варианты CLU и PICALM, связанные с болезнью Альцгеймера. Нат Жене. 2009;41(10):1088–1093. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Нат Жене. 2009;41(10):1088–1093. [Бесплатная статья PMC] [PubMed] [Google Scholar]
23. Lambert JC, Heath S, Even G, et al. Исследователи Европейской инициативы по борьбе с болезнью Альцгеймера Исследование ассоциации всего генома выявляет варианты в CLU и CR1, связанные с болезнью Альцгеймера. Нат Жене. 2009 г.;41(10):1094–1099. [PubMed] [Google Scholar]
24. Сешадри С., Фитцпатрик А.Л., Икрам М.А. и др. Консорциум CHARGE. Консорциум ГЕРАД1; Консорциум EADI1 Полногеномный анализ генетических локусов, связанных с болезнью Альцгеймера. ДЖАМА. 2010;303(18):1832–1840. [Бесплатная статья PMC] [PubMed] [Google Scholar]
25. Холлингворт П., Гарольд Д., Симс Р. и др. Инициатива нейровизуализации болезни Альцгеймера. ЗАРЯД консорциум. Консорциум EADI1 Общие варианты в ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 и CD2AP связаны с болезнью Альцгеймера. Нат Жене. 2011;43(5):429–435. 10.1038/нг.803. [Бесплатная статья PMC] [PubMed] [Google Scholar]
26. Беттенс К., Слигерс К. , Ван Брокховен К. Текущее состояние молекулярной генетики болезни Альцгеймера: от прошлого к настоящему и к будущему. Хум Мол Жене. 2010;19(R1):R4–R11. [Бесплатная статья PMC] [PubMed] [Google Scholar]
, Ван Брокховен К. Текущее состояние молекулярной генетики болезни Альцгеймера: от прошлого к настоящему и к будущему. Хум Мол Жене. 2010;19(R1):R4–R11. [Бесплатная статья PMC] [PubMed] [Google Scholar]
27. Yerbury JJ, Poon S, Meehan S, et al. Внеклеточный шаперон кластерин влияет на образование амилоида и токсичность, взаимодействуя с префибриллярными структурами. FASEB J. 2007;21(10):2312–2322. [PubMed] [Академия Google]
28. Киршбаум Л., Бозас С.Е., Уокер И.Д. SP-40,40, белок, участвующий в контроле пути комплемента, обладает уникальным набором дисульфидных мостиков. ФЭБС лат. 1992;297(1-2):70–76. [PubMed] [Google Scholar]
29. Тамбисетти М., Симмонс А., Велаюдхан Л. и соавт. Связь концентрации кластерина в плазме с тяжестью, патологией и прогрессированием болезни Альцгеймера. Арх генерал психиатрия. 2010;67(7):739–748. [Бесплатная статья PMC] [PubMed] [Google Scholar]
30. Calero M, Rostagno A, Frangione B, Ghiso J. Кластерин и болезнь Альцгеймера. Субклеточная биохимия. 2005; 38: 273–29.8. [PubMed] [Google Scholar]
2005; 38: 273–29.8. [PubMed] [Google Scholar]
31. Braskie MN, Jahanshad N, Stein JL, et al. Распространенный вариант риска болезни Альцгеймера в гене CLU влияет на микроструктуру белого вещества у молодых людей. Дж. Нейроски. 2011;31(18):6764–6770. [Бесплатная статья PMC] [PubMed] [Google Scholar]
32. Krych-Goldberg M, Atkinson JP. Структурно-функциональные отношения рецептора комплемента типа 1. Immunol Rev. 2001;180:112–122. [PubMed] [Google Scholar]
33. Zanjani H, Finch CE, Kemper C, et al. Активация комплемента на очень ранней стадии болезни Альцгеймера. Альцгеймер Dis Assoc Disord. 2005;19(2): 55–66. [PubMed] [Google Scholar]
34. Eikelenboom P, Veerhuis R, Scheper W, Rozemuller AJ, van Gool WA, Hoozemans JJ. Значение нейровоспаления в понимании болезни Альцгеймера. J Neural Transm. 2006;113(11):1685–1695. [PubMed] [Google Scholar]
35. Rogers J, Li R, Mastroeni D, et al. Периферический клиренс бета-амилоидного пептида за счет комплементарной С3-зависимой адгезии к эритроцитам. Нейробиол Старение. 2006; 27(12):1733–1739. [PubMed] [Академия Google]
Нейробиол Старение. 2006; 27(12):1733–1739. [PubMed] [Академия Google]
36. Brouwers N, Van Cauwenberghe C, Engelborghs S, et al. Риск болезни Альцгеймера, связанный с изменением числа копий в рецепторе комплемента 1, увеличивающим сайты связывания C3b/C4b. Мол Психиатрия. 2011 15 марта; [Epub перед печатью] [Бесплатная статья PMC] [PubMed] [Google Scholar]
37. Тебар Ф., Боландер С.К., Соркин А. Белок лимфоидной миелоидной лейкемии (CALM) сборки клатрина: локализация в ямках, покрытых эндоцитами, взаимодействие с клатрин и влияние гиперэкспрессии на клатрин-опосредованный трафик. Мол Биол Селл. 1999;10(8):2687–2702. [Бесплатная статья PMC] [PubMed] [Google Scholar]
38. Koo EH, Squazzo SL. Доказательства того, что производство и высвобождение бета-амилоидного белка происходит по эндоцитарному пути. Дж. Биол. Хим. 1994;269(26):17386–17389. [PubMed] [Google Scholar]
39. Carey RM, Balcz BA, Lopez-Coviella I, Slack BE. Ингибирование динамин-зависимого эндоцитоза увеличивает выделение эктодомена белка-предшественника амилоида и снижает образование бета-амилоидного белка. BMC клеточная биология. 2005; 6:30. [Бесплатная статья PMC] [PubMed] [Google Scholar]
BMC клеточная биология. 2005; 6:30. [Бесплатная статья PMC] [PubMed] [Google Scholar]
40. Harel A, Wu F, Mattson MP, Morris CM, Yao PJ. Доказательства CALM в управлении торговлей VAMP2. Движение. 2008;9(3):417–429. [PubMed] [Google Scholar]
41. Сакамуро Д., Эллиотт К.Дж., Векслер-Рейя Р., Прендергаст Г.К. BIN1 представляет собой новый белок, взаимодействующий с MYC, обладающий свойствами супрессора опухолей. Нат Жене. 1996;14(1):69–77. [PubMed] [Google Scholar]
42. Wigge P, McMahon HT. Семейство белков амфифизина и их роль в эндоцитозе в синапсе. Тренды Нейроси. 1998;21(8):339–344. [PubMed] [Google Scholar]
43. Meunier B, Quaranta M, Daviet L, Hatzoglou A, Leprince C. Мембранно-тубулирующий потенциал амфифизина 2/BIN1 зависит от цитоплазматического линкерного белка 170, связывающего микротрубочки (CLIP- 170) Eur J Cell Biol. 2009;88(2):91–102. [PubMed] [Google Scholar]
44. Танака Н., Абэ-Дохмаэ С., Ивамото Н., Йокояма С. Роль АТФ-связывающего кассетного переносчика A7 в гомеостазе холестерина и системе защиты хозяина. J Атеросклеротический тромб. 2011;18(4):274–281. [PubMed] [Академия Google]
J Атеросклеротический тромб. 2011;18(4):274–281. [PubMed] [Академия Google]
45. Ikeda Y, Abe-Dohmae S, Munehira Y, et al. Посттранскрипционная регуляция ABCA7 человека и его функция для зависимого от апоА-I высвобождения липидов. Biochem Biophys Res Commun. 2003;311(2):313–318. [PubMed] [Google Scholar]
46. Langmann T, Mauerer R, Zahn A, et al. Профилирование экспрессии с обратной транскрипцией и ПЦР в реальном времени полного надсемейства АТФ-связывающих кассетных транспортеров человека в различных тканях. Клин Хим. 2003;49(2):230–238. [PubMed] [Google Scholar]
47. Jehle AW, Gardai SJ, Li S, et al. АТФ-связывающий кассетный переносчик A7 усиливает фагоцитоз апоптотических клеток и связанную с ним передачу сигналов ERK в макрофагах. Джей Селл Биол. 2006;174(4):547–556. [Бесплатная статья PMC] [PubMed] [Google Scholar]
48. Лян Ю., Бакли Т.Р., Ту Л., Лэнгдон С.Д., Теддер Т.Ф. Структурная организация кластера генов MS4A человека на хромосоме 11q12. Иммуногенетика. 2001;53(5):357–368. [PubMed] [Google Scholar]
2001;53(5):357–368. [PubMed] [Google Scholar]
49. Tateno H, Li H, Schur MJ, et al. Различные эндоцитарные механизмы CD22 (Siglec-2) и Siglec-F отражают роль в клеточной передаче сигналов и врожденном иммунитете. Мол Селл Биол. 2007;27(16):5699–5710. [Бесплатная статья PMC] [PubMed] [Google Scholar]
50. Crocker PR, Paulson JC, Varki A. Siglecs и их роль в иммунной системе. Нат Рев Иммунол. 2007;7(4):255–266. [PubMed] [Академия Google]
51. Dustin ML, Olszowy MW, Holdorf AD, et al. Новый адапторный белок управляет формированием паттерна рецепторов и полярностью цитоскелета в контактах Т-клеток. Клетка. 1998;94(5):667–677. [PubMed] [Google Scholar]
52. Lynch DK, Winata SC, Lyons RJ, et al. Комплекс кортактин-CD2-ассоциированный белок (CD2AP) обеспечивает новую связь между эндоцитозом рецептора эпидермального фактора роста и актиновым цитоскелетом. Дж. Биол. Хим. 2003;278(24):21805–21813. [PubMed] [Google Scholar]
53. Пимпликар С.В., Никсон Р.А., Робакис Н. К., Шен Дж., Цай Л.Х. Амилоиднезависимые механизмы в патогенезе болезни Альцгеймера. Дж. Нейроски. 2010;30(45):14946–14954. [Бесплатная статья PMC] [PubMed] [Google Scholar]
К., Шен Дж., Цай Л.Х. Амилоиднезависимые механизмы в патогенезе болезни Альцгеймера. Дж. Нейроски. 2010;30(45):14946–14954. [Бесплатная статья PMC] [PubMed] [Google Scholar]
54. McGeer EG, McGeer PL. Врожденный иммунитет при болезни Альцгеймера: модель местных воспалительных реакций. Мол Интерв. 2001;1(1):22–29. [PubMed] [Google Scholar]
55. Bates KA, Verdile G, Li QX, et al. Механизмы клиренса бета-амилоидного пептида болезни Альцгеймера: последствия для терапевтического дизайна и диагностических тестов. Мол Психиатрия. 2009;14(5):469–486. [PubMed] [Google Scholar]
56. Бласко И., Штампфер-Кунтчев М., Робачер П., Веерхуис Р., Эйкеленбум П., Грубек-Лебенштейн Б. Как хроническое воспаление может повлиять на мозг и способствовать развитию болезни Альцгеймера в пожилом возрасте : роль микроглии и астроцитов. Стареющая клетка. 2004;3(4):169–176. [PubMed] [Google Scholar]
57. Rowe JA, Molds JM, Newbold CI, Miller LHP. Розеткообразование P. falciparum, опосредованное паразитарным вариантом мембранного белка эритроцитов и рецептором комплемента 1. Природа. 1997;388(6639):292–295. [PubMed] [Google Scholar]
Природа. 1997;388(6639):292–295. [PubMed] [Google Scholar]
58. Dietschy JM, Turley SD. Метаболизм холестерина в головном мозге. Карр Опин Липидол. 2001;12(2):105–112. [PubMed] [Google Scholar]
59. Puglielli L, Tanzi RE, Kovacs DM. Болезнь Альцгеймера: связь с холестерином. Нат Нейроски. 2003;6(4):345–351. [PubMed] [Академия Google]
60. Miwa Y, Takiuchi S, Kamide K, et al. Полиморфизм вставки/делеции в гене кластерина влияет на уровень липидов в сыворотке крови и толщину интима-медиа сонных артерий у японских женщин с гипертензией. Biochem Biophys Res Commun. 2005;331(4):1587–1593. [PubMed] [Google Scholar]
61. Ishikawa Y, Akasaka Y, Ishii T, et al. Распределение и синтез аполипопротеина J в атеросклеротической аорте. Артериосклеры Тромб Васк Биол. 1998;18(4):665–672. [PubMed] [Google Scholar]
62. Танака Н., Абэ-Дохмаэ С., Ивамото Н., Йокояма С. Роль АТФ-связывающего кассетного переносчика A7 в гомеостазе холестерина и системе защиты хозяина. J Атеросклеротический тромб. 2011;18(4):274–281. [PubMed] [Академия Google]
J Атеросклеротический тромб. 2011;18(4):274–281. [PubMed] [Академия Google]
63. Мак Дж.Т., Таунсенд Д.М., Белянски В., Тью К.Д. Транспортер ABCA2: внутриклеточная роль в транспортировке и метаболизме холестерина, полученного из ЛПНП, и соединений, связанных со стеролами. Curr Drug Metab. 2007;8(1):47–57. [Бесплатная статья PMC] [PubMed] [Google Scholar]
64. Brody DL, Magnoni S, Schwetye KE, et al. Динамика бета-амилоида коррелирует с неврологическим статусом в поврежденном мозге человека. Наука. 2008;321(5893):1221–1224. [Бесплатная статья PMC] [PubMed] [Google Scholar]
65. Shankar GM, Li S, Mehta TH, et al. Димеры белка бета-амилоида, выделенные непосредственно из мозга больных Альцгеймером, нарушают синаптическую пластичность и память. Нат Мед. 2008;14(8):837–842. [Бесплатная статья PMC] [PubMed] [Google Scholar]
66. Селкое Д.Дж. Болезнь Альцгеймера. Колд Спринг Харб Перспект Биол. 2011;3(7):ii, a004457. [Бесплатная статья PMC] [PubMed] [Google Scholar]
67. Бартл М.М., Лукенбах Т., Бергнер О., Ульрих О., Кох-Брандт С. Множественные рецепторы опосредуют апоJ-зависимый клиренс клеточного мусора в непрофессиональные фагоциты. Разрешение ячейки опыта. 2001;271(1):130–141. [PubMed] [Google Scholar]
Бартл М.М., Лукенбах Т., Бергнер О., Ульрих О., Кох-Брандт С. Множественные рецепторы опосредуют апоJ-зависимый клиренс клеточного мусора в непрофессиональные фагоциты. Разрешение ячейки опыта. 2001;271(1):130–141. [PubMed] [Google Scholar]
68. Bell RD, Sagare AP, Friedman AE, et al. Транспортные пути для клиренса бета-амилоидного пептида болезни Альцгеймера человека и аполипопротеинов E и J в центральной нервной системе мыши. J Cereb Blood Flow Metab. 2007;27(5):909–918. [Бесплатная статья PMC] [PubMed] [Google Scholar]
69. Stevens B, Allen NJ, Vazquez LE, et al. Классический каскад комплемента обеспечивает элиминацию синапсов ЦНС. Клетка. 2007;131(6):1164–1178. [PubMed] [Google Scholar]
70. Rademakers R, Rovelet-Lecrux A. Последние данные о молекулярной генетике деменции. Тренды Нейроси. 2009;32(8):451–461. [Бесплатная статья PMC] [PubMed] [Google Scholar]
71. Rohrer JD, Guerreiro R, Vandrovcova J, et al. Наследуемость и генетика лобно-височной долевой дегенерации. Неврология. 2009 г.;73(18):1451–1456. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Неврология. 2009 г.;73(18):1451–1456. [Бесплатная статья PMC] [PubMed] [Google Scholar]
72. van der Zee J, Sleegers K, Van Broeckhoven C. Приглашенная статья: спектр дегенерации лобно-височной доли болезни Альцгеймера. Неврология. 2008;71(15):1191–1197. [PubMed] [Google Scholar]
73. Ballatore C, Lee VM, Trojanowski JQ. Тау-опосредованная нейродегенерация при болезни Альцгеймера и родственных расстройствах. Нат Рев Нейроски. 2007;8(9):663–672. [PubMed] [Google Scholar]
74. Чен-Плоткин А.С., Ли В.М., Трояновский JQ. TAR ДНК-связывающий белок 43 при нейродегенеративных заболеваниях. Нат Рев Нейрол. 2010;6(4):211–220. [Бесплатная статья PMC] [PubMed] [Google Scholar]
75. Mackenzie IR, Rademakers R, Neumann M. TDP-43 и FUS при боковом амиотрофическом склерозе и лобно-височной деменции. Ланцет Нейрол. 2010;9(10):995–1007. [PubMed] [Google Scholar]
76. Urwin H, Josephs KA, Rohrer JD, et al. Патология FUS Consortium FReJA определяет большинство случаев тау- и TDP-43-негативной лобно-височной долевой дегенерации. Акта Нейропатол. 2010;120(1):33–41. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Акта Нейропатол. 2010;120(1):33–41. [Бесплатная статья PMC] [PubMed] [Google Scholar]
77. Coppola G, Karydas A, Rademakers R, et al. Исследование экспрессии генов в периферической крови выявляет мутации програнулина. Энн Нейрол. 2008;64(1):92–96. [Бесплатная статья PMC] [PubMed] [Google Scholar]
78. Ghidoni R, Benussi L, Glionna M, Franzoni M, Binetti G. Низкие уровни програнулина в плазме предсказывают мутации програнулина при дегенерации лобно-височной доли. Неврология. 2008;71(16):1235–1239. [PubMed] [Google Scholar]
79. Sleegers K, Brouwers N, Van Damme P, et al. Сывороточный биомаркер ассоциированной с програнулином лобно-височной долевой дегенерации. Энн Нейрол. 2009;65(5):603–609. [PubMed] [Google Scholar]
80. Geser F, Martinez-Lage M, Robinson J, et al. Клинико-патологический континуум мультисистемных протеинопатий TDP-43. Арх Нейрол. 2009 г.;66(2):180–189. [Бесплатная статья PMC] [PubMed] [Google Scholar]
81. Barmada SJ, Skibinski G, Korb E, Rao EJ, Wu JY, Finkbeiner S. Цитоплазматическая неправильная локализация TDP-43 токсична для нейронов и усиливается связанной с ней мутацией. с семейным боковым амиотрофическим склерозом. Дж. Нейроски. 2010;30(2):639–649. [Бесплатная статья PMC] [PubMed] [Google Scholar]
Цитоплазматическая неправильная локализация TDP-43 токсична для нейронов и усиливается связанной с ней мутацией. с семейным боковым амиотрофическим склерозом. Дж. Нейроски. 2010;30(2):639–649. [Бесплатная статья PMC] [PubMed] [Google Scholar]
82. Weihl CC. Валозин-содержащая белок-ассоциированная лобно-височная долевая дегенерация: клиника, патологические особенности и патогенез. Curr Alzheimer Res. 2011;8(3):252–260. [Бесплатная статья PMC] [PubMed] [Google Scholar]
83. Urwin H, Ghazi-Noori S, Collinge J, Isaacs A. Роль CHMP2B в лобно-височной деменции. Биохим Сок Транс. 2009; 37 (часть 1): 208–212. [PubMed] [Google Scholar]
84. Lee JA, Liu L, Gao FB. Дефекты аутофагии способствуют нейродегенерации, вызванной дисфункциональным ESCRT-III. Аутофагия. 2009;5(7):1070–1072. [PubMed] [Google Scholar]
85. Van Deerlin VM, Sleiman PM, Martinez-Lage M, et al. Распространенные варианты 7p21 связаны с лобно-височной долевой дегенерацией с включениями TDP-43. Нат Жене.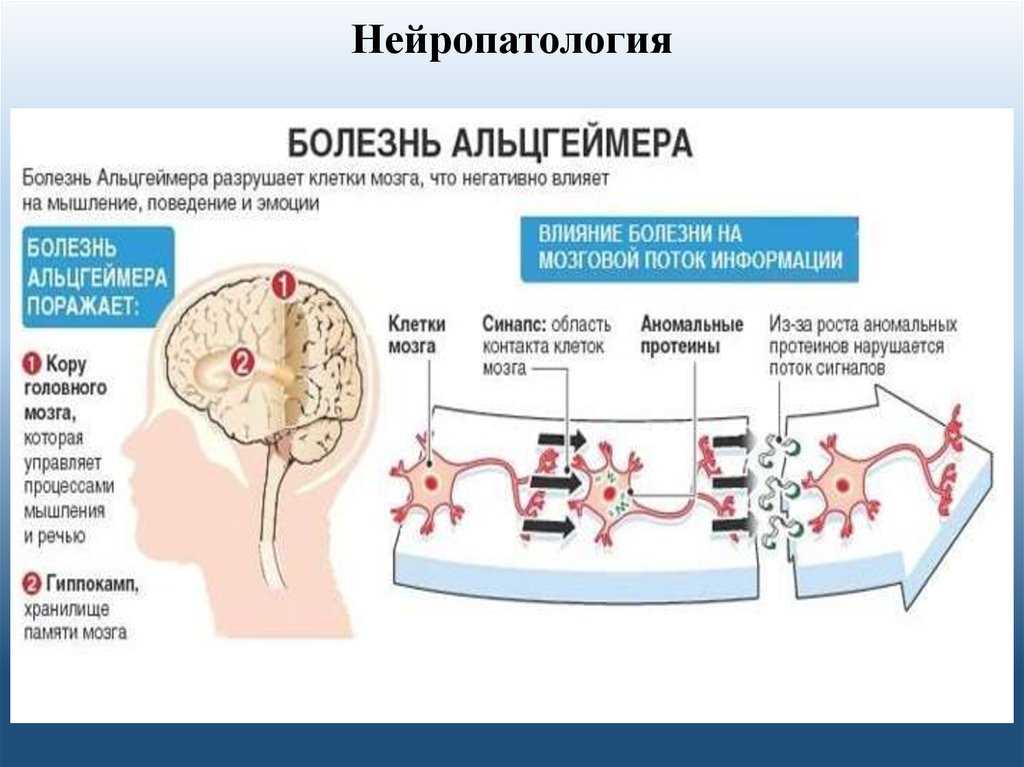 2010;42(3):234–239. [Бесплатная статья PMC] [PubMed] [Google Scholar]
2010;42(3):234–239. [Бесплатная статья PMC] [PubMed] [Google Scholar]
86. Finch N, Carrasquillo MM, Baker M, et al. TMEM106B регулирует уровни програнулина и пенетрантность FTLD у носителей мутации GRN. Неврология. 2011;76(5):467–474. [Бесплатная статья PMC] [PubMed] [Google Scholar]
87. Бонифати В. Последние достижения в генетике деменции с тельцами Леви. Curr Neurol Neurosci Rep. 2008;8(3):187–189. [PubMed] [Google Scholar]
88. Clark LN, Kartsaklis LA, Wolf Gilbert R, et al. Ассоциация мутаций глюкоцереброзидазы с деменцией с тельцами Леви. Арх Нейрол. 2009 г.;66(5):578–583. [Бесплатная статья PMC] [PubMed] [Google Scholar]
89. Браун К., Мастрианни Дж.А. Прионные болезни. J Geriatr Psychiatry Neurol. 2010;23(4):277–298. [PubMed] [Google Scholar]
90. Mead S, Poulter M, Uphill J, et al. Генетические факторы риска варианта болезни Крейтцфельдта-Якоба: полногеномное ассоциативное исследование. Ланцет Нейрол. 2009;8(1):57–66. [Бесплатная статья PMC] [PubMed] [Google Scholar]
91. Angot E, Steiner JA, Hansen C, Li JY, Brundin P. Являются ли синуклеинопатии прионоподобными расстройствами? Ланцет Нейрол. 2010;9(11): 1128–1138. [PubMed] [Google Scholar]
Angot E, Steiner JA, Hansen C, Li JY, Brundin P. Являются ли синуклеинопатии прионоподобными расстройствами? Ланцет Нейрол. 2010;9(11): 1128–1138. [PubMed] [Google Scholar]
92. Kim J, Holtzman DM. Лекарственное средство. Прионоподобное поведение бета-амилоида. Наука. 2010;330(6006):918–919. [PubMed] [Google Scholar]
93. Фрост Б., Даймонд М.И. Прионоподобные механизмы при нейродегенеративных заболеваниях. Нат Рев Нейроски. 2010;11(3):155–159. [Бесплатная статья PMC] [PubMed] [Google Scholar]
94. Chabriat H, Joutel A, Dichgans M, Tournier-Lasserve E, Bousser MG. Кадасил. Ланцет Нейрол. 2009;8(7):643–653. [PubMed] [Академия Google]
95. Росс CA, Табризи SJ. Болезнь Гентингтона: от молекулярного патогенеза к клиническому лечению. Ланцет Нейрол. 2011;10(1):83–98. [PubMed] [Google Scholar]
96. Wild EJ, Mudanohwo EE, Sweeney MG, et al. Фенокопии болезни Гентингтона клинически и генетически гетерогенны. Мов Беспорядок. 2008;23(5):716–720. [PubMed] [Google Scholar]
97. Стеванин Г., Брайс А. Спиноцеребеллярная атаксия 17 (SCA17) и мозжечок, подобный болезни Гентингтона 4 (HDL4). 2008;7(2):170–178. [PubMed] [Академия Google]
Стеванин Г., Брайс А. Спиноцеребеллярная атаксия 17 (SCA17) и мозжечок, подобный болезни Гентингтона 4 (HDL4). 2008;7(2):170–178. [PubMed] [Академия Google]
98. Гарсия-Аросена Д., Хагерман П.Дж. Прогресс в понимании молекулярной основы FXTAS. Хум Мол Жене. 2010;19(R1):R83–R89. [Бесплатная статья PMC] [PubMed] [Google Scholar]
99. Leehey MA. Fragile X-ассоциированный тремор/синдром атаксии: клинический фенотип, диагностика и лечение. J Исследовательская Мед. 2009;57(8):830–836. [Бесплатная статья PMC] [PubMed] [Google Scholar]
100. Renton AE, Majounie E, Waite A, et al. Расширение гексануклеотидного повтора в C9ORF72 является причиной хромосомы 9.p21-связанный БАС-ЛВД. Нейрон. 2011;72(2):257–268. [Бесплатная статья PMC] [PubMed] [Google Scholar]
101. DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Расширенный гексануклеотидный повтор GGGGCC в некодирующей области C9ORF72 вызывает FTD и ALS, сцепленные с хромосомой 9p. Нейрон. 2011;72(2):245–256. [Бесплатная статья PMC] [PubMed] [Google Scholar]
102.