Атрофия мышечная это: Врач СПбГПМУ рассказала об опыте лечения спинальной мышечной атрофии
Почему у людей бывает СМА?
Спинальная мышечная атрофия – это заболевание, при котором повреждается ген SMN1. В результате мутации у человека не вырабатывается белок, который нужен для выживания мотонейронов. Мотонейроны — важная часть спинного мозга человека, от них по длинным нервам идут сигналы к скелетным мышцам организма. Если мотонейрон получает недостаточно белка для своего питания, то он быстро устает. Ему приходится работать и за себя, и «за того парня». Многие мотонейроны просто гибнут от «усталости», — нагрузка на оставшиеся возрастает, и в результате они работают с чудовищной перегрузкой. От усталости они не могут посылать сигналы к скелетным мышцам, благодаря которым человек ходит, сидит, лежит, глотает или дышит. Мышцы остаются без работы, потому что их никто ничего не просит сделать, и постепенно отмирают.
Дефицит важного белка, который не вырабатывает ген SMN1, приводит к атрофии (потере жизнеспособности и уменьшению размера) мышц, и в результате человек постепенно теряет способность ходить, удерживать и управлять собственным телом, самостоятельно сидеть, есть, глотать и дышать.
К счастью, у человека есть еще ген SMN2, который дублирует SMN1. Он производит нефункциональный белок и небольшое количество нормального белка, который позволяет мотонейронам человека выживать на очень голодном пайке. В результате человек со СМА сохраняет какую то часть своих двигательных, глотательных и дыхательных функций.
От чего зависит тип СМА и тяжесть болезни?
Если максимально упростить картину, то у человека может быть разное число копий гена SMN2. Теоретически, чем больше у пациента со СМА таких копий, которые вырабатывают маленькое количество нужного мотонейронам белка, тем легче будет протекать заболевание, человек сохранит больше жизненно важных функций и дольше проживет. На практике есть слишком много нюансов, чтобы напрямую связывать тяжесть болезней с числом копий SMN2.
При этом у людей признаки СМА появляются в разное время: от первых дней жизни до взрослого возраста.
Существует четыре типа спинальной мышечной атрофии. Чем раньше начинается заболевание, тем тяжелее оно протекает. В 95 % случаев причиной СМА становится делеция (выпадение) участка генетического кода.
Чем раньше начинается заболевание, тем тяжелее оно протекает. В 95 % случаев причиной СМА становится делеция (выпадение) участка генетического кода.
По международным стандартам типы СМА различаются несколькими важнейшими признаками:
Болезнь Верднига-Гофманна (СМА 1), возраст дебюта заболевания от 0 до 6 месяцев, максимальная (двигательная) функция у таких детей: сами не могут сидеть. При естественном течении заболевания 92 % этих деток погибает до возраста двух лет.
Болезнь Дубовица (СМА 2). Дебют заболевания происходит с 6 до 18 месяцев. Такие пациенты никогда не могут самостоятельно стоять, они могут самостоятельно сидеть. Продолжительность их жизни может быть больше двух лет
Болезнь Кугельберга-Велендер (СМА 3). Дебют заболевания происходит позже 18 месяцев, такие пациенты развиваются и нормально ходят, но в какой-то момент заболевание начинает прогрессировать. Они перестают самостоятельно передвигаться и садятся. Хотя продолжительность жизни у них тоже сильно ограничена, но они могут прожить несколько десятилетий.
Четвертый тип мы почти не видим в детской клинике. Такие пациенты дебютируют в подростковом и взрослом возрасте, они могут потерять способность к самостоятельному передвижению. Продолжительность жизни — десятилетия.
Тип СМА | Возраст дебюта заболевания | Максимальная функция | Средняя продолжитель-ность жизни | Типичные проявления |
Тип 1 Болезнь Верднига-Гофманна | 0-6 месяцев | Не сидит | < 2-х лет | Глубокая слабость и гипотония, трудности контроля головы, слабый крик и кашель, трудности с глотанием и выведением мокроты, осложненное течение ОРВИ, развитие дыхательной недостаточности, высокие риски аспирационной пневмонии, очень высокая скорость прогрессирования заболевания |
Тип 2 Болезнь Дубовица | 6-18 месяцев | Не стоит | > 2-х лет | Задержка моторного развития и набора веса, слабый кашель, тремор рук, контрактуры, сколиоз и деформация грудной клетки. |
Тип 3 Болезнь Кугельберга-Веландер | Старше 18 месяцев | Стоит и ходит | Десятилетия | Мышечная слабость различной степени выраженности, крампи (судороги в икроножных мышцах), контрактуры и гипермобильность суставов, потеря способности ходить по мере прогрессирования заболевания |
Тип 4 Поздний тип | В подростковом или взрослом возрасте | Могут потерять способность к самостоятельному передвижению | Десятилетия | Прогрессирующая проксимальная мышечная слабость, снижение сухожильных рефлексов, фасцикуляции |
Чуть более подробно и наглядно об особенностях разных типов СМА можно посмотреть по ссылке
Почему рождается ребенок со СМА?
Спинальная мышечная атрофия — это генетическое заболевание. Каждый сороковой житель планеты — здоровый носитель СМА. Чтобы в семье родился ребенок со СМА, нужно, чтобы такими носителями были оба родителя. В этом случае в 25% случаях ребенок родится здоровым, в 50% случаях он получает мутацию гена SMN1 от папы или от мамы и тоже становится просто носителем, а еще в 25% случаях он наследует мутацию гена SMN1 от обоих родителей и рождается с этой болезнью.
Чтобы в семье родился ребенок со СМА, нужно, чтобы такими носителями были оба родителя. В этом случае в 25% случаях ребенок родится здоровым, в 50% случаях он получает мутацию гена SMN1 от папы или от мамы и тоже становится просто носителем, а еще в 25% случаях он наследует мутацию гена SMN1 от обоих родителей и рождается с этой болезнью.
Отметим, что у здоровых носителей мутации гена SMN1 есть один полноценный ген и один ген с мутацией. Наличие одного полноценного гена достаточно для выработки необходимого количества нормального белка и правильного функционирования мотонейронов. Так что носитель от человека без поломки этого гена отличается лишь потенциальным риском рождения ребенка со СМА.
Более наглядно это показано на схеме
К сожалению, в реальности генетические особенности человека срабатывают не по математическому принципу.
Кандидат медицинских наук, ведущий врач-генетик ООО «Центра Генетики и Репродуктивной Медицины «Генетико» Наталья Ветрова описывает реальную практику появления детей со СМА в семьях, где есть два носителя:
«В семье могут быть все здоровыми, могут быть все носителями, могут быть все больными, могут через один.
Закон 25-50-25 действует на очень больших числах. В реальных семьях люди с мутацией гена SMN1 не могут иметь такое большое потомство, чтобы было распределение так, как у нас на картинке».
 Закон 25-50-25 действует на очень больших числах. В реальных семьях люди с мутацией гена SMN1 не могут иметь такое большое потомство, чтобы было распределение так, как у нас на картинке».
Закон 25-50-25 действует на очень больших числах. В реальных семьях люди с мутацией гена SMN1 не могут иметь такое большое потомство, чтобы было распределение так, как у нас на картинке».Кто и как ставит диагноз «Спинальная мышечная атрофия»
Поставить клинический диагноз СМА врач может после неврологического осмотра пациента с проверкой рефлексов и мышечного тонуса. Еще нужно провести внешний осмотр пациента и сделать биохимический тест.
После этого человек должен пройти еще несколько тестов. Генетическое исследование должно подтвердить или опровергнуть первоначальный клинический диагноз.
Тесты также нужны для уточнения прогноза заболевания и уточнения носительства мутаций гена SMN1 у родителей ребенка.
Подробнее о назначении каждого теста можно узнать, посмотрев на изображение.
Лучший тест на сегодня — MLPA. Он подтверждает причину СМА в 95 % случаев. Для оставшихся 5 % нужно тест секвенирование гена SMN1 (он нужен, если первый тест не подтвердил диагноз, а у пациента есть клинические проявления СМА).
Еще один тест, который может назначить врач, это панель «Нервно-мышечные заболевания» (НМЗ). Он нужен в случае, когда ни MPLA ни секвенирование не смогли найти причину клинических проявлений, которые позволяют врачу заподозрить СМА или другое нервно — мышечное заболевание.
СМА и беременность
Если в семье уже есть ребенок со СМА или один из будущих родителей — носитель или пациент со СМА, необходимо заранее провести несколько тестов .
Если два основных генетических исследования (MPLA и секвенирование) показывают, что второй родитель тоже носитель мутации гена SMN1, нужно решить, каким образом запланировать и провести беременность, чтобы минимизировать риск появления ребенка со СМА.
Если супружеская пара выбирает естественный путь для зачатия, то она должна пройти пренатальную инвазивную диагностику после десятой недели беременности. Для этого прокалывают живот и делают забор материала у плода. В современных условиях процедура достаточно безопасна и для будущей мамы, и для плода. Существует минимальный риск выкидыша, но он гораздо ниже, чем вероятность рождения ребенка со СМА.
Существует минимальный риск выкидыша, но он гораздо ниже, чем вероятность рождения ребенка со СМА.
Если супруги предпочитают ЭКО, то они могут пойти двумя путями:
или из нескольких зародышей отобрать здорового, или воспользоваться услугами донора — неносителя СМА. В этом случае донора нужно дополнительно исследовать на наличие мутации в гене SMN1.
При отборе эмбрионов риск возможного повреждения каждого эмбриона стремится к нулю. Отобранный здоровый эмбрион замораживают и подсаживают в матку.
Более подробно о беременности при СМА можно узнать, посмотрев запись вебинара Натальи Ветровой.
Проиграть видео
Патогенетическая терапия при СМА
В настоящее время в мире есть два уже одобренных препарата для лечения СМА — Нусинерсен (Спинраза) и AVXS -101 (Золгенсма). Ожидается, что в ближайшее время на рынке появится третий препарат RG7916 (Рисдиплам).
Нусинерсен предназначен для пожизненного приема, в случае с Золгенсма речь идет об однократной дозе, но оба этих препарата работают неодинаково. Один решает проблему мышечной слабости, другой — препарат, который модифицирует генные клетки в целом.
Один решает проблему мышечной слабости, другой — препарат, который модифицирует генные клетки в целом.
Главная задача этих препаратов — восполнить дефицит белка из-за не работающего гена SMN1. О проведенных клинических испытаниях препаратов и о последних новостях лекарственной терапии при СМА можно прочитать в разделе Клинические исследования и лекарства.
Краткий обзор о принципах работы патогенетической терапии при СМА можно прочитать по ссылке.
Эта статья для медицинских работников. Мы не рекомендуем пациентам использовать данные из нее самостоятельно. За консультациями необходимо обратится к врачу. Информация, предоставленная в статье, может относиться к лекарственным препаратам, отпускаемым по рецепту.
Лечение спинальной мышечной атрофии (CMA)
В больнице «Ихилов» теперь предоставляется новаторская терапия СМА (спинальной мышечной атрофии) — заболевания, которое до сих пор считалось неизлечимым. Речь идет о лечении препаратом Спинраза (Spinraza). Лекарство получило утверждение Управления по контролю качества пищевых продуктов и лекарственных средств в США.
Речь идет о лечении препаратом Спинраза (Spinraza). Лекарство получило утверждение Управления по контролю качества пищевых продуктов и лекарственных средств в США.
Узнать цены
Содержание
-
1 . Что такое болезнь Спинальная мышечная атрофия (CMA)?
-
2 . Что такое лечение Spinraza?
-
3 .
Лечение мышечной дистрофии в Медицинском центре Ихилов в Израиле:
 Лечение мышечной дистрофии в Медицинском центре Ихилов в Израиле:
Лечение мышечной дистрофии в Медицинском центре Ихилов в Израиле:
Что такое болезнь Спинальная мышечная атрофия (CMA)?
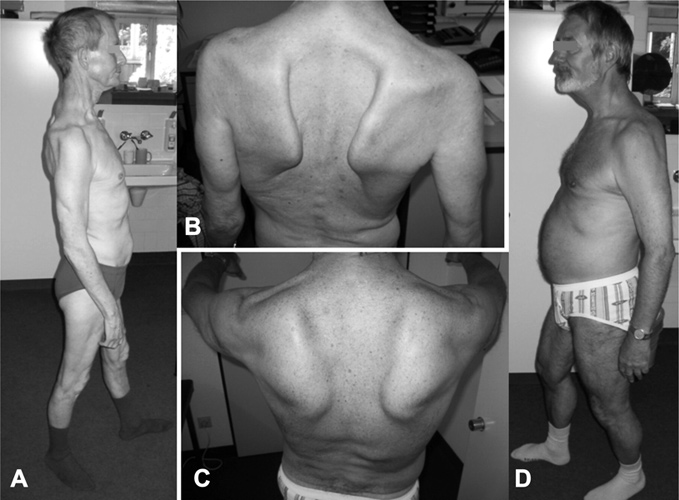
СМА (спинальная мышечная атрофия) — редкое генетическое заболевание, передающееся наследственным путем в тех случаях, когда оба родителя являются носителями. Недуг проявляется в слабости мышц, вплоть до паралича. В большинстве случаев в процесс вовлекаются и дыхательные мышцы. Заболевание развивается вследствие генетического дефекта, приводящего к атрофии определенного рода нервных клеток передних рогов спинного мозга. Встречаемость заболевания составляет от 1:6000 до 1:11000. Диагноз ставится обычно в младенческом или в раннем дошкольном возрасте. Чем раньше появляются первые симптомы, тем более неблагоприятное течение заболевания.
Что такое лечение Spinraza?
Препарат Спинраза впервые позволяет проводить терапию, воздействующую на генетическом уровне, а именно, стимулирующую ген SMN2, который выполняет ту же функцию, что и отсутствующий у больных ген SMN1.
Для обеспечения доставки препарата в нервную систему практикуется интратекальное введение.
Спинраза доказала свою эффективность в качестве терапии, спасающей жизнь при СМА. Речь идет о первом препарате, показавшем столь исключительные результаты. Лекарство предотвращает прогрессирование заболевания и улучшает функциональный статус больных. Некоторые из пациентов, получавших Спинразу, вновь обрели способность самостоятельно ходить. Специалисты характеризуют воздействие лекарства как «чудодейственное».
Лечение мышечной дистрофии в Медицинском центре Ихилов в Израиле:
В Тель-авивском медицинском центре действует специальная многопрофильная клиника по ведению пациентов с СМА.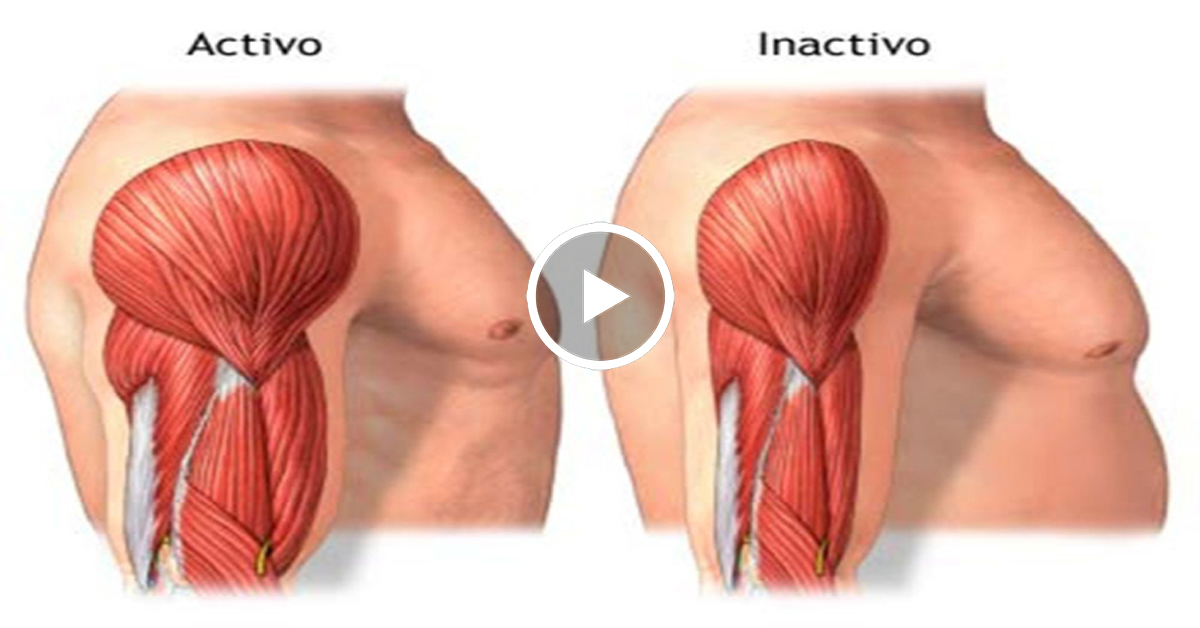 Руководит ею профессор Паталь, специалист с мировым именем. Пациенты абмулатории — дети и молодые люди.
Руководит ею профессор Паталь, специалист с мировым именем. Пациенты абмулатории — дети и молодые люди.
Первичное освидетельствование производится детским неврологом, специализирующимся на нервно-мышечных заболеваниях.
Вопросы и ответы
Отделения клиники
Информация об атрофии мышц | Гора Синай
Атрофия мышц; истощение; Атрофия мышц
Мышечная атрофия – это истощение (истончение) или потеря мышечной ткани.
С возрастом люди могут терять от 20 до 40 процентов мышечной массы, а вместе с ней и силы. Ученые обнаружили, что основная причина, по которой люди теряют мышцы, заключается в том, что они перестают заниматься повседневными делами, требующими мышечной силы, а не только потому, что стареют.
Ученые обнаружили, что основная причина, по которой люди теряют мышцы, заключается в том, что они перестают заниматься повседневными делами, требующими мышечной силы, а не только потому, что стареют.
Мышечная атрофия – это уменьшение размера и истощение мышечной ткани. Мышцы, которые теряют иннервацию, могут атрофироваться и просто чахнуть.
Причины
Существует три типа мышечной атрофии: физиологическая, патологическая и нейрогенная.
Физиологическая атрофия вызвана недостаточным использованием мышц. Этот тип атрофии часто можно обратить вспять с помощью упражнений и лучшего питания. Больше всего страдают люди, которые:
- Имеют сидячую работу, имеют проблемы со здоровьем, которые ограничивают движение или снижают уровень активности
- Прикованы к постели
- Не могут двигать конечностями из-за инсульта или другого заболевания головного мозга
- Находятся в месте, где отсутствует гравитация, например, во время космических полетов
Патологическая атрофия наблюдается при старении, голодании и таких заболеваниях, как болезнь Кушинга (из-за приема слишком большого количества лекарств, называемых кортикостероидами).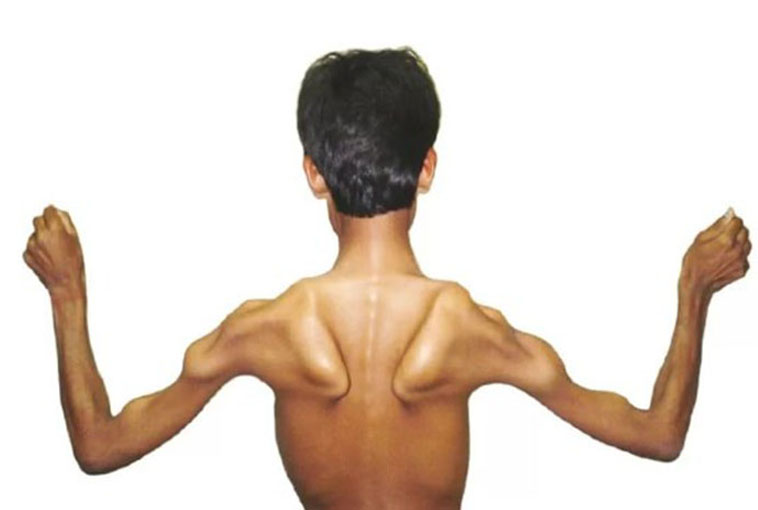
Нейрогенная атрофия является наиболее тяжелым типом мышечной атрофии. Это может быть вызвано травмой или заболеванием нерва, который соединяется с мышцей. Этот тип мышечной атрофии имеет тенденцию возникать более внезапно, чем физиологическая атрофия.
Примеры заболеваний, поражающих нервы, управляющие мышцами:
- Боковой амиотрофический склероз (БАС или болезнь Лу Герига)
- Повреждение одного нерва, такое как синдром запястного канала
- Синдром Гийена-Барре
- Повреждение нерва, вызванное травмой, диабетом, токсинами или алкоголем
- Полиомиелит (полиомиелит)
- Травма спинного мозга
Хотя люди могут адаптироваться к мышечной атрофии, даже незначительная мышечная атрофия вызывает некоторую потерю подвижности или силы.
Другие причины мышечной атрофии могут включать:
- Ожоги
- Длительная кортикостероидная терапия
- Недоедание
- Мышечная дистрофия и другие заболевания мышц
- Остеоартрит
- Ревматоидный артрит
Уход на дому
Программа упражнений может помочь в лечении мышечной атрофии. Упражнения могут включать упражнения в бассейне для снижения мышечной нагрузки и другие виды реабилитации. Ваш лечащий врач может рассказать вам больше об этом.
Упражнения могут включать упражнения в бассейне для снижения мышечной нагрузки и другие виды реабилитации. Ваш лечащий врач может рассказать вам больше об этом.
Люди, которые не могут активно двигать одним или несколькими суставами, могут выполнять упражнения с использованием скоб или шин.
Когда обращаться к медицинскому работнику
Свяжитесь со своим поставщиком медицинских услуг, чтобы записаться на прием, если у вас необъяснимая или длительная потеря мышечной массы. Это часто можно увидеть, если сравнить одну руку, руку или ногу с другой.
Чего ожидать от врача Посетите
Медицинский работник проведет медицинский осмотр и спросит о вашей истории болезни и симптомах, в том числе:
- Когда началась мышечная атрофия?
- Становится хуже?
- Какие еще симптомы у вас есть?
Медицинский работник осмотрит ваши руки и ноги и измерит размер мышц.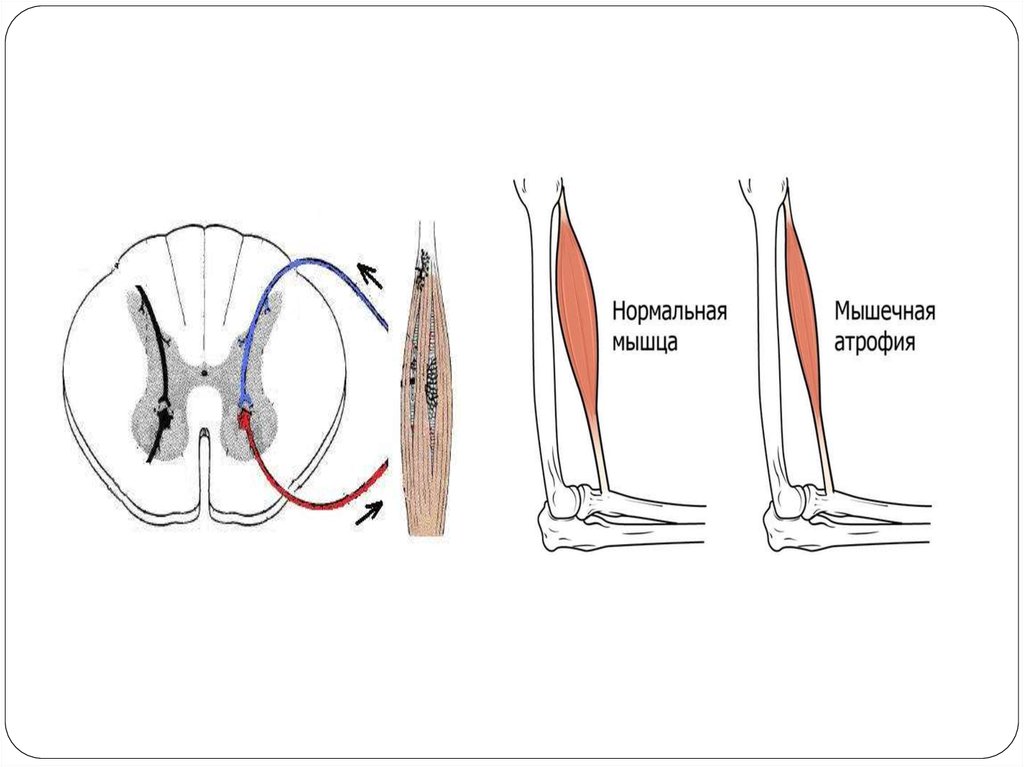 Это может помочь определить, какие нервы поражены.
Это может помочь определить, какие нервы поражены.
Тесты, которые могут быть выполнены, включают:
- Анализы крови
- КТ
- Электромиография (ЭМГ)
- МРТ
- Биопсия мышц или нервов
- Исследования проводимости нервов
- Рентген Лечение, ультразвук и в некоторых случаях физиотерапия, ультразвук
Болл Дж. В., Дейнс Дж. Э., Флинн Дж. А., Соломон Б. С., Стюарт Р. В. Костно-мышечной системы. В: Болл Дж. В., Дейнс Дж. Э., Флинн Дж. А., Соломон Б. С., Стюарт Р. В., ред. Руководство Зайделя по физикальному обследованию . 10-е изд. Сент-Луис, Миссури: Elsevier; 2023:глава 22.
Selcen D. Заболевания мышц. В: Goldman L, Schafer AI, ред. Медицина Голдман-Сесил . 26-е изд. Филадельфия, Пенсильвания: Elsevier; 2020: глава 393.
Последнее рассмотрение: 09.11.2021
Рецензировал: Джозеф В. Кампеллоне, доктор медицинских наук, отделение неврологии, Медицинская школа Купера при Университете Роуэна, Камден, Нью-Джерси. Обзор предоставлен VeriMed Healthcare Network. Также рассмотрены Дэвидом Зивом, доктором медицины, MHA, медицинским директором, Брендой Конауэй, редакционным директором, и A.D.A.M. Редакционная коллегия.
Спинальная мышечная атрофия: типы СМА
Спинальная мышечная атрофия — это генетическое заболевание, поражающее нервную систему и мышцы. Люди с этим заболеванием испытывают мышечную слабость и истощение.
Спинальная мышечная атрофия (СМА) — это генетическое заболевание, которым страдает 1 человек из 10 000. Это ухудшает способность человека контролировать свои движения мышц. Хотя у всех со СМА есть генная мутация, начало, симптомы и прогрессирование заболевания значительно различаются.
По этой причине СМА часто подразделяют на четыре типа. Различные генные мутации могут вызывать другие редкие формы СМА.
Различные генные мутации могут вызывать другие редкие формы СМА.
Читайте дальше, чтобы узнать о различных типах SMA.
Все четыре основных типа СМА возникают в результате дефицита белка, называемого SMN, что означает «выживание двигательных нейронов». Моторные нейроны — это нервные клетки в спинном мозге, которые посылают сигналы нашим мышцам.
Когда мутация происходит в обеих копиях гена SMN1 , это приводит к дефициту белка SMN. Если белок SMN вырабатывается мало или вообще не вырабатывается, это приводит к проблемам с двигательной функцией.
Гены, соседствующие с SMN1 , называемые генами SMN2 , сходны по структуре с генами SMN1 . Иногда они могут помочь компенсировать дефицит белка SMN. Но количество SMN2 генов колеблется от человека к человеку. Таким образом, тип СМА зависит от того, сколько генов SMN2 есть у человека, чтобы компенсировать мутацию гена SMN1 .
Если у человека со СМА, связанной с хромосомой 5, больше копий SMN2 , они могут производить больше рабочего белка SMN.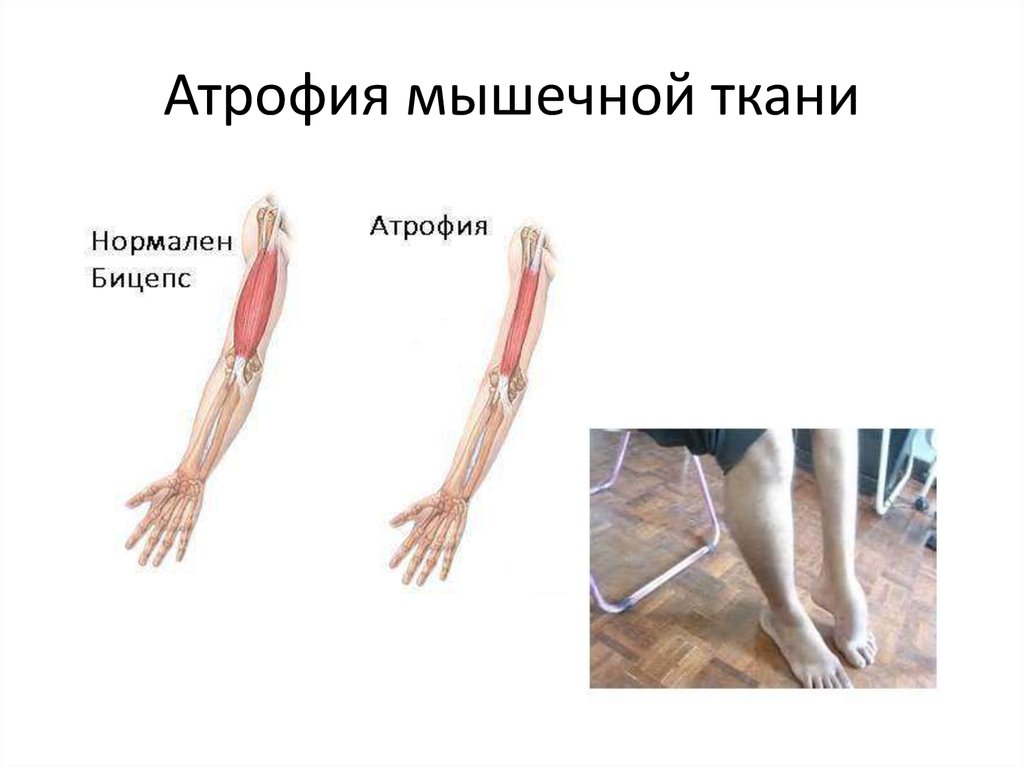 В свою очередь, их SMA будет мягче с более поздним началом, чем у тех, у кого меньше копий гена SMN2 .
В свою очередь, их SMA будет мягче с более поздним началом, чем у тех, у кого меньше копий гена SMN2 .
СМА является аутосомно-рецессивным заболеванием, поэтому, чтобы унаследовать это заболевание, человек должен получить две мутировавшие копии гена, по одной от каждого родителя. Человек только с одной копией мутировавшего гена называется носителем. У носителей нет никаких симптомов СМА, но они могут передать мутировавший ген своим детям.
Тип 0, также называемый пренатальной СМА, является наиболее тяжелой формой СМА. Он характеризуется мышечной слабостью и истощением, которые появляются во время внутриутробного развития и присутствуют при рождении. Врожденные пороки сердца также часто встречаются у людей со СМА 0 типа.
Прогноз
Младенцы со СМА типа 0 часто умирают в течение первых шести месяцев жизни.
СМА типа 1 также называется СМА с инфантильным началом или болезнью Верднига-Гоффмана. Он затрагивает около 60% людей со СМА. Обычно этот тип возникает из-за наличия только одной или двух копий SMN2 Ген, по одному на каждой хромосоме 5. Более половины новых диагнозов СМА относятся к типу 1.
Более половины новых диагнозов СМА относятся к типу 1.
Когда появляются симптомы
У детей со СМА типа 1 симптомы проявляются в течение первых шести месяцев после рождения. Среднее начало заболевания происходит в возрасте около 2,5 месяцев.
Симптомы
Симптомы СМА 1 типа могут включать:
- слабость, дряблость рук и ног (гипотония)
- неспособность сидеть
- гастроэзофагеальный рефлюкс (ГЭРБ)
- проблемы с движением, глотанием и дыханием
- проблемы с поднятием головы
- потеря костной массы, переломы и аномалии позвоночника, такие как сколиоз
Перспективы
Благодаря новым достижениям в лечении и уходе продолжительность жизни пациентов со СМА 1 типа растет. Однако многие дети с этой формой СМА не доживают до двухлетнего возраста.
СМА типа 2 также называется промежуточной СМА и поражает около 30% людей с этим заболеванием. В целом, у многих людей со СМА 2 типа есть три SMN2 ген.
Когда появляются симптомы
Симптомы СМА 2 типа обычно проявляются в возрасте 18 месяцев.
Симптомы
Симптомы СМА типа 2, как правило, менее выражены, чем симптомы СМА типа 1. Они могут включать:
- слабые руки и ноги
- трудности при ходьбе без посторонней помощи
- аномальное искривление позвоночника
- слабые подергивания дыхательных мышц или аномальные движения
Перспективы
СМА 2 типа может сократить продолжительность жизни, но большинство людей со СМА 2 типа доживают до зрелого возраста. Люди со СМА 2 типа часто пользуются инвалидной коляской. Им также может понадобиться оборудование, которое поможет им лучше дышать ночью.
СМА типа 3 также называют ювенильной СМА, легкой формой СМА или болезнью Кугельберга-Веландера. Он затрагивает около 10% людей со СМА. Симптомы этого типа более вариабельны. Люди со СМА 3 типа обычно имеют от трех до четырех генов SMN2 .
Когда появляются симптомы
Симптомы появляются после 18-месячного возраста. Обычно его диагностируют в возрасте 3 лет, но точный возраст начала заболевания варьируется. У некоторых могут не проявляться симптомы до раннего взросления.
Обычно его диагностируют в возрасте 3 лет, но точный возраст начала заболевания варьируется. У некоторых могут не проявляться симптомы до раннего взросления.
Симптомы
Люди со СМА 3 типа обычно могут стоять и ходить самостоятельно, но с возрастом они могут потерять способность ходить. Другие симптомы могут включать:
- затруднения при вставании из положения сидя
- проблемы с равновесием
- трудности при подъеме по ступенькам или беге
- мышечная усталость
- усиление слабости с течением времени
Перспективы
обычно изменяют продолжительность жизни человека, но люди с этим типом могут иметь повышенный риск избыточного веса. Их кости также могут стать слабыми и легко ломаться.
СМА 4 типа также называется СМА с поздним началом. Это затрагивает около 1% людей с этим заболеванием. Люди со СМА 4 типа имеют от трех до пяти генов SMN2 и могут производить достаточное количество белка SMN. Тип 4 является наименее распространенным из четырех типов.
Тип 4 является наименее распространенным из четырех типов.
Когда появляются симптомы
Симптомы СМА 4 типа обычно проявляются в раннем взрослом возрасте, обычно после 35 лет.
Симптомы
СМА 4 типа могут постепенно ухудшаться с течением времени. Люди с этим типом СМА обычно могут ходить самостоятельно. Симптомы могут включать: 9Слабость в руках и ногах . Большинство людей со СМА 4 типа могут жить и работать независимо и обычно не нуждаются во вспомогательных устройствах для передвижения.
Эти типы СМА встречаются редко и вызываются генными мутациями, отличными от тех, которые влияют на белок SMN.
- Спинальная мышечная атрофия с респираторным дистресс-синдромом (SMARD) — очень редкая форма СМА, вызванная мутацией гена IGHMBP2 . SMARD диагностируется у младенцев и вызывает серьезные проблемы с дыханием.
- Болезнь Кеннеди, или спинально-бульбарная мышечная атрофия (СБМА), — это редкий вид СМА, которым обычно страдают только мужчины. Он часто начинается в возрасте от 20 до 40 лет. Симптомы включают слабость лицевых мышц, трудности с глотанием, тремор рук, мышечные судороги, слабость в конечностях и подергивания. Хотя это также может вызвать трудности при ходьбе в более позднем возрасте, этот тип СМА обычно не влияет на ожидаемую продолжительность жизни.
- Дистальная СМА — редкая форма, вызванная мутациями в одном из многих генов, включая UBA1 , DYNC1h2 и GARS . Поражает нервные клетки спинного мозга. Симптомы обычно начинаются в подростковом возрасте и включают судороги или слабость и истощение мышц. Это не влияет на продолжительность жизни.
 Он часто начинается в возрасте от 20 до 40 лет. Симптомы включают слабость лицевых мышц, трудности с глотанием, тремор рук, мышечные судороги, слабость в конечностях и подергивания. Хотя это также может вызвать трудности при ходьбе в более позднем возрасте, этот тип СМА обычно не влияет на ожидаемую продолжительность жизни.
Он часто начинается в возрасте от 20 до 40 лет. Симптомы включают слабость лицевых мышц, трудности с глотанием, тремор рук, мышечные судороги, слабость в конечностях и подергивания. Хотя это также может вызвать трудности при ходьбе в более позднем возрасте, этот тип СМА обычно не влияет на ожидаемую продолжительность жизни.Ниже приведены некоторые из наиболее часто задаваемых вопросов о SMA.
Насколько распространена спинальная мышечная дистрофия?
Подсчитано, что 1 из каждых 10 000 новорожденных имеет СМА.
Как диагностируется спинальная мышечная дистрофия?
Врачи могут назначить несколько тестов для диагностики СМА. Наиболее распространенным тестом является генетический тест для поиска мутации, вызывающей СМА. Другие тесты могут включать электромиографию (ЭМГ) для измерения электрической активности мышц и исследование нервной проводимости для измерения того, насколько хорошо нервы способны посылать сигналы. Анализ крови на креатинкиназу (КК) также может быть сделан для выявления высоких уровней КК, что может быть признаком повреждения мышц. В редких случаях может быть выполнена биопсия мышц для поиска изменений в структуре мышечных волокон.
Наиболее распространенным тестом является генетический тест для поиска мутации, вызывающей СМА. Другие тесты могут включать электромиографию (ЭМГ) для измерения электрической активности мышц и исследование нервной проводимости для измерения того, насколько хорошо нервы способны посылать сигналы. Анализ крови на креатинкиназу (КК) также может быть сделан для выявления высоких уровней КК, что может быть признаком повреждения мышц. В редких случаях может быть выполнена биопсия мышц для поиска изменений в структуре мышечных волокон.
Какие существуют варианты лечения спинальной мышечной атрофии?
Лекарства от СМА не существует, но есть методы лечения, которые могут помочь справиться с симптомами и улучшить качество жизни. Доступны лекарства, модифицирующие заболевание, такие как нусинерсен (Спинраза), рисдиплам (Еврисди) и онсемноген абепарвовек-ксиоли (Золгенсма). Эти процедуры могут помочь улучшить мышечную силу, функцию и качество дыхания.
Физиотерапия, трудотерапия и корсеты могут помочь сохранить мышечную силу и функцию. Существуют также препараты, которые могут помочь уменьшить воспаление и замедлить прогрессирование заболевания. Некоторым людям также может потребоваться респираторная поддержка. Клинические испытания новых методов лечения СМА продолжаются.
Существуют также препараты, которые могут помочь уменьшить воспаление и замедлить прогрессирование заболевания. Некоторым людям также может потребоваться респираторная поддержка. Клинические испытания новых методов лечения СМА продолжаются.
Каков прогноз для человека со спинальной мышечной атрофией?
Прогноз для людей со СМА зависит от их типа СМА. Люди со СМА 1 и 2 типов обычно имеют более короткую продолжительность жизни, в то время как люди со СМА 3 и 4 типов обычно имеют среднюю продолжительность жизни. Однако СМА может вызвать такие осложнения, как проблемы с дыханием и слабость костей, что может сократить продолжительность жизни.
Существует четыре различных типа СМА, связанной с хромосомой 5, примерно соответствующие возрасту, в котором появляются симптомы. Тип зависит от числа SMN2 генов человек должен помочь компенсировать мутацию в гене SMN1 .
В общем, более ранний возраст начала заболевания означает меньшее количество копий SMN2 и большее влияние на двигательную функцию.